

Effective hemostasis depends on an adequate number of functional platelets as well as an adequate concentration and activity of cell surface and plasma proteins (coagulation factors).
Traditionally, the coagulation cascade has been modeled in terms of discrete extrinsic and intrinsic pathways leading to a common pathway (see figure and table ):
In the intrinsic pathway, factor XIIa (FXIIa) and FXIa interact to activate FIX to FIXa. FIXa then combines with FVIIIa to form a complex (intrinsic tenase) that activates FX to FXa.
In the extrinsic pathway, factor VIIa and tissue factor directly activate FX. The TF-FVIIa complex (extrinsic tenase) can also activate FIX of the intrinsic pathway (so-called “alternate pathway”).
In the common pathway, FXa associates with its cofactor FVa to activate FII (prothrombin) to FIIa (thrombin). Thrombin cleaves FI (fibrinogen) to soluble fibrin (FIa), which is then crosslinked to insoluble fibrin by FXIIIa.
Components of Blood Coagulation Reactions
Component | Description |
|---|---|
Tissue factor (TF) | TF is a transmembrane glycoprotein receptor found in extravascular tissues, including organ capsules and the adventitia of blood vessel walls. It is constitutively expressed on fibroblasts and, on cellular activation, on vascular smooth muscle cells, monocytes, and neutrophils. The TF-bearing cells and platelet surfaces act as the main cellular surfaces for assembly of the procoagulant complexes. It is exposed to flowing blood during injury or inflammation, binds FVIIa, and initiates the extrinsic coagulation pathway. |
Fibrinogen (FI) | Fibrinogen is proteolytically cleaved to form fibrin, producing a firm clot. |
Prothrombin (FII) | Prothrombin is proteolytically cleaved to form thrombin. Thrombin, in turn, converts fibrinogen to fibrin; activates soluble FV, FVIII, FXI, and FXIII; and binds to thrombomodulin to activate PC. |
FV | FV is principally synthesized in the liver and is activated to FVa by FXa and thrombin. FXa associates with its cofactor FVa to form prothrombinase complexes that activate FII (prothrombin) to FIIa (thrombin). |
FVII | Factor VII binding to TF results in activation to Factor VIIa. Factor VIIa bound to TF on the cell surface activates Factor IX to Factor IXa and Factor X to Factor Xa. |
FVIII | FVIII and von Willebrand factor (vWF) circulate in plasma as a noncovalent bimolecular complex. Upon activation by thrombin, FVIIIa dissociates from the FVIII-vWF complex to interact with FIXa. |
FIX | FIXa on platelets and TF-bearing cells binds with FVIIIa, assembling the FIXa-FVIIIa complex (intrinsic tenase). The FIXa-FVIIIa complex activates FX. |
FX | FX is activated to FXa, the enzyme of the prothrombinase complex that cleaves prothrombin to thrombin. |
FXI | FXIIa and FXIa interact to activate FIX. |
FXII | Once bound to TF, the proenzyme FVII is activated to the enzyme FVIIa. The TF-FVIIa complex (extrinsic tenase) activates FX, triggering the intrinsic coagulation pathway (in vitro). |
FXIII | When activated by thrombin, FXIIIa cross-links adjacent fibrin monomers to strengthen and stabilize fibrin clots. |
Protein C (PC) | PC is a serine protease that is the precursor to the protease referred to as activated protein C (PCa). |
Protein S (PS) | PS is serine protease that is a nonenzymatic cofactor that enhances activity of PCa. |
Thrombomodulin (TM) | TM is a cell surface receptor for thrombin, mainly expressed on undamaged endothelium. The antithrombic effect of TM involves removal of thrombin escaped from sites of vascular injury. |
However, the simplified conceptualization of extrinsic and intrinsic coagulation pathways no longer is considered a sufficient platform for discussing homeostatic mechanisms for achieving and maintaining hemostatic balance.

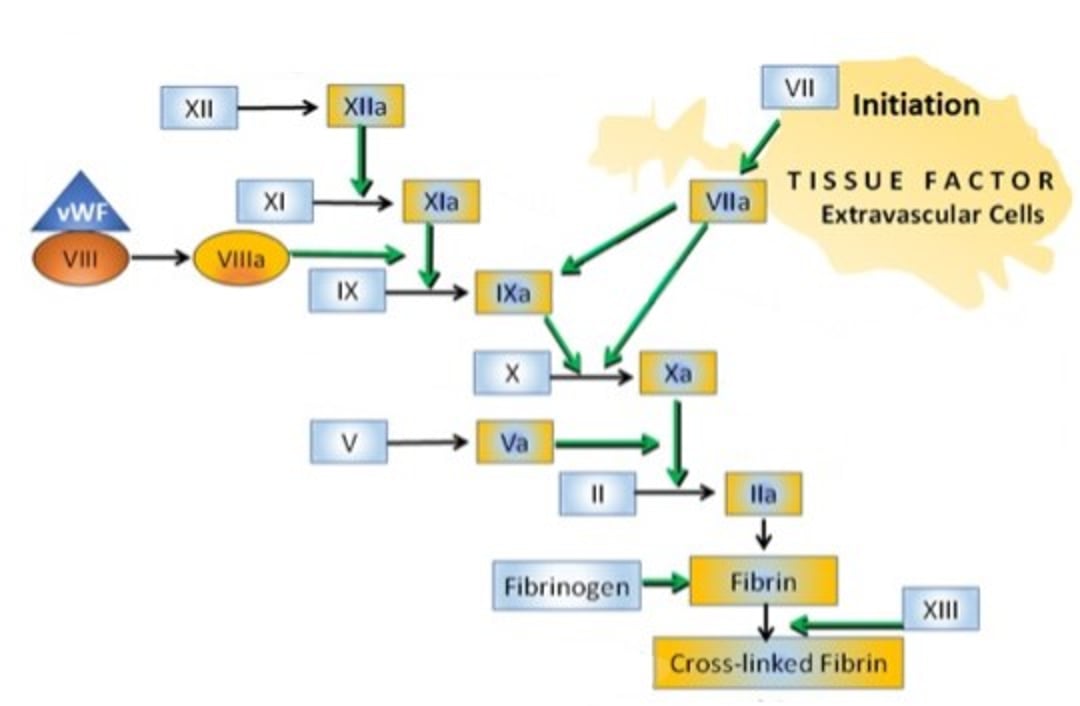
Courtesy of Dr. Sharon Center.
The coagulation process can be considered in terms of primary and secondary hemostatic mechanisms followed by fibrinolytic and anticoagulant modifications.
Primary hemostasis involves an initiating vascular endothelial injury that exposes subendothelial collagen and releases von Willebrand factor (vWF) from the endothelium, forming a stringlike platform as a “landing site” for circulating platelets. Platelets also bind to other extracellular matrix proteins at sites of injury. On exposure to subendothelial collagen, platelets undergo transformational activation and aggregation.
Along with microvascular vasospasm at the injury site, these responses rapidly control bleeding. Adequacy of this response can be crudely assessed with a consistently performed buccal mucosal bleeding time.
However, primary hemostatic mechanisms provide only a temporary solution for hemorrhage control. The platelet plug must be fortified by a resilient meshwork of stabilized fibrin that is only achieved by secondary hemostasis.
Secondary hemostasis begins with an initiation process that starts the cascade of coagulation factor activation that will eventually generate thrombin. While described in discrete steps (see ), this is a complex process of dynamic interactions overlapping in a chaotic continuum.
The initiating step is exposure of tissue factor (TF)-bearing cells to a site of injury.
Tissue factor is a transmembrane protein receptor and cofactor for FVII, expressed by extravascular cells. Being highly expressed on pericytes enveloping arterioles and venules and by adventitial cells surrounding large vessels, TF-associated cells provide a so-called hemostatic envelope.
Once bound to TF, the proenzyme FVII is rapidly activated to the enzyme FVIIa, which in turn catalyzes the activation of FX and FIX. Thereafter, FXa localized on the TF-bearing cell interacts with its cofactor FVa, generating a small quantity of thrombin.
Perivascular binding of FVII/FVIIa to the TF-bearing cells occurs even in the absence of vascular injury (coagulative proteins extravasate into the extravascular space, eventually diffusing into lymph). FIX, FX, and thrombin are produced on TF-bearing cells at all times, allowing their prompt use.
Activated factors are generally separated from other coagulative proteins by the vascular wall. The large dimensions of platelets and FVIII bound to vWF, however, restricts their entry to the extravascular compartment until vascular injury.
Activated platelets release a partially activated platelet-specific form of FV that promptly combines with FXa on TF-bearing cells forming prothrombinase complexes. Interaction of the small amount of thrombin on TF-bearing cells with the FVIII-vWF complex ignites the cascade of factor activation that will evolve the thrombin that will laminate the original hemostatic platelet plug.
The initiation stage is followed by an amplification stage, engaging maximal platelet activation and the activation of additional coagulation cofactors on the platelet surface. This priming reaction fuels additional bursts of thrombin formation on the platelet surface (ie, activating FV, FVIII, and FXI). FIXa on platelets and TF-bearing cells binds with FVIIIa assembling the FIXa-FVIIIa (intrinsic tenase) complexes.
Finally, a propagation phase generates the burst of thrombin formation needed for effective hemostasis. Once the platelet tenase complex is assembled, plasma FX is activated to FXa on the platelet surface, where it associates with FVa, forming prothrombinase complexes. This interaction initiates a final burst of thrombin generation.
The PT assay assesses adequacy of procoagulants involved in the initiation phase of secondary coagulation. The aPTT assay assesses adequacy of procoagulants involved in platelet-surface mediated thrombin generation during the propagation phase.
Coagulation Inhibitors in Animals
The coagulation inhibitors antithrombin (AT) and tissue factor pathway inhibitor (TFPI) are critical for limiting and localizing FXa activity and thrombin generation to a site of injury. FXa on a cell surface is relatively protected from inhibition by AT and TFPI whereas FXa in solution is rapidly inactivated.
Both TFPI and AT are localized to endothelial cell surfaces, where they thwart inappropriate diffusion of activated factors or inappropriate formation of factors distant to the initial site of injury. AT binds to heparin sulfates on endothelial cells; its deficiency poses a notable risk for thrombosis.
Antithrombin is a serine protease inhibitor that physiologically inactivates thrombin (FIIa) and FXa and, to a lesser extent, FIXa, FXIa, and FXIIa; tissue plasminogen activator (tPA); urokinase; trypsin; plasmin; and kallikrein (see figure ).

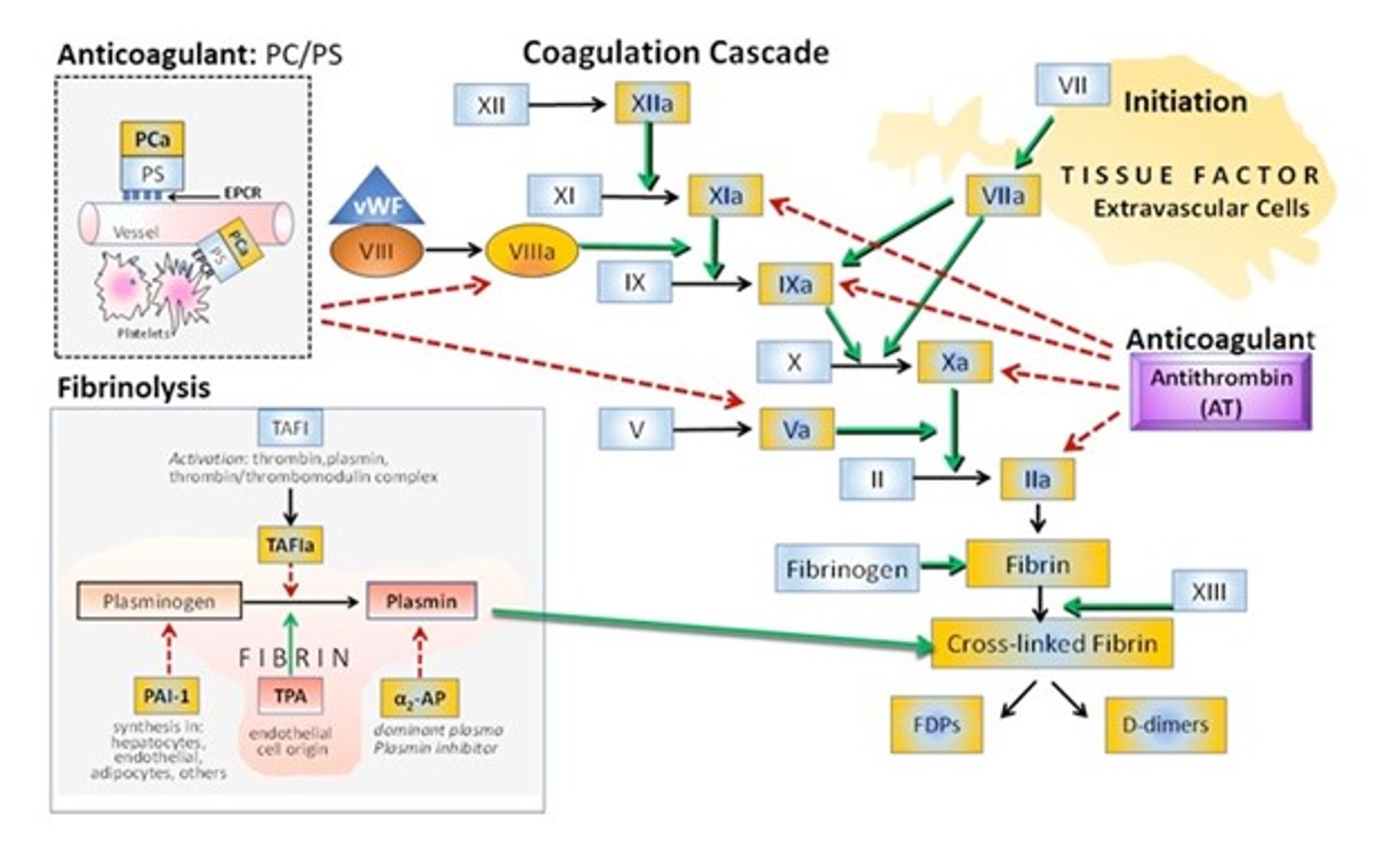
Courtesy of Dr. Sharon Center,
The anticoagulative activity of AT is dramatically accelerated by conformational change induced by heparin binding that increases exposure of its molecularly reactive binding center. This conformational change converts AT from a slow to a rapid inactivator of coagulation factors by ~1,000-fold.
Endogenous heparan sulfates attached to vascular endothelium physiologically activate AT and localize its inhibitory activity to the intraluminal vascular endothelium. Because the therapeutic effect of heparin involves this molecular interaction, heparin resistance may be realized in some patients with hypercoagulable status and venous thrombi associated with low AT activity.
Additional to its essential anticoagulant effect, AT also decreases platelet adhesion to fibrinogen, imparts an anti-inflammatory effect to endothelium (decreased IL-6, IL-8 release), and increases vascular release of prostacyclin, which mediates vascular relaxation and vasodilation and inhibits platelet aggregation.
Low AT activity particularly increases risk for venous thromboembolism.
Protein C (PC) and protein S (PS) are vitamin K–dependent serine proteases. PC is the precursor to the protease referred to as activated protein C (PCa). PS is a nonenzymatic cofactor that enhances activity of PCa. PC exerts its antithrombotic effect when bound to a specific endothelial protein C receptor (EPCR); this binding enhances its activation and localizes its effect to endothelial surfaces (see figure ).
Thrombomodulin (TM) is a cell surface receptor for thrombin, mainly expressed on undamaged endothelium. The antithrombic effect of TM involves removal of thrombin escaped from sites of vascular injury.
Formation of a thrombin-TM complex activates PC localized to platelet surfaces by EPCR. Here, PCa binds with its PS cofactor, inactivating FVa on endothelium, thereby disabling thrombin production. The PCa-PS complex thwarts thrombin generation distant to the site of vascular injury but does not interfere with the process of clot formation. PS has additional antithrombotic effect in enhancing the inhibition of FXa by TFPI and by directly interacting with FVa, inhibiting unwarranted prothrombinase complex activity.
PCa also exhibits potent cytoprotective and anti-inflammatory properties and indirectly functions as a fibrinolytic, inactivating plasminogen-activator inhibitor-1 (PAI-1) and reducing release of thrombin activatable fibrinolysis inhibitor (TAFI). Deficiency of either PC or PS can provoke thrombotic tendencies.
Fibrinolysis in Animals
The fibrinolytic system is a checkpoint for control of inappropriate or unrestricted clot formation. First, it degrades clots formed within intact vasculature, protecting against intravascular thrombosis. Secondly, it gradually dissipates the hemostatic clot during wound healing and tissue repair.
Similar to the anticoagulant proteins, the fibrinolytic system is localized to an appropriate surface and not systemically mobilized or expressed. Localization is restricted by binding of fibrinolytic moieties to specific cell receptors and to fibrin.
Most fibrinolytic proteins are synthesized by the liver (see figure ).
The major fibrinolytic enzyme is plasmin, formed from a circulating zymogen plasminogen by plasminogen activators (tissue plasminogen activator [tPA] or urinary plasminogen activator [uPA or urokinase]). While plasminogen is synthesized in the liver, tPA is synthesized by endothelial cells in response to ischemia or hypoxia and thrombin.
There is also an inhibitor that modulates tPA, synthesized by hepatocytes, endothelial cells, adipocytes, and other cell types. This is denoted as plasminogen activator inhibitor-1 (PAI-1). Increased elaboration of PAI-1 increases risk for thrombus formation whereas insufficient PAI-1 increases risk for critical hemorrhage.
Both the generation and activity of plasmin is restricted to the surface of the fibrin polymer, where tPA bound to fibrin activates plasminogen. Thus, fibrin formation and deposition is linked to its own degradation.
Fibrinolysis is also inhibited by alpha2-antiplasmin (alpha2-AP) and thrombin-activatable fibrinolysis inhibitor (TAFI), proteases also synthesized in the liver. Alpha2-AP is the major plasma inhibitor of plasmin in the circulation and also may inactivate trypsin, elastase, and PC. Alpha2-AP circulates in both a free form and bound to plasminogen.
TAFI circulates as a proenzyme and is converted to a potent attenuator of the fibrinolytic system upon activation by thrombin, plasmin, or the thrombin-thrombomodulin complex. Functionally, TAFI downregulates fibrinolysis by removing C-terminal lysine residues from partially degraded fibrin, thereby preventing the upregulation of plasminogen binding and activation. TAFI is considered an important homeostatic link between thrombin formation and fibrinolysis, enabling fine synchronization between these two arms of hemostasis.
Clot breakdown products include D-dimer and FDPs (fibrinogen and fibrin degradation products, which are circulating fragments of fibrinogen and soluble fibrin).


