



Hematologic changes in patients with liver disease are generally nonspecific and overlap with many other systemic disorders. The exception is that icteric plasma may be a hallmark of liver disease in the absence of severe anemia, which would implicate a hemolytic process.
Red Blood Cell Effects
Depending on the severity and underlying cause, liver disease may be associated with nonregenerative or regenerative anemia of variable severity.
Severe or acute anemia of any origin can impact hepatocytes secondary to hypoxia, causing membrane alterations that may be associated with transient release of hepatic transaminases (ALT, AST). An example of a severe impact occurs in some dogs with immune-mediated hemolytic anemia, where agglutinated RBCs in hepatic sinusoids interfere with transhepatic perfusion.

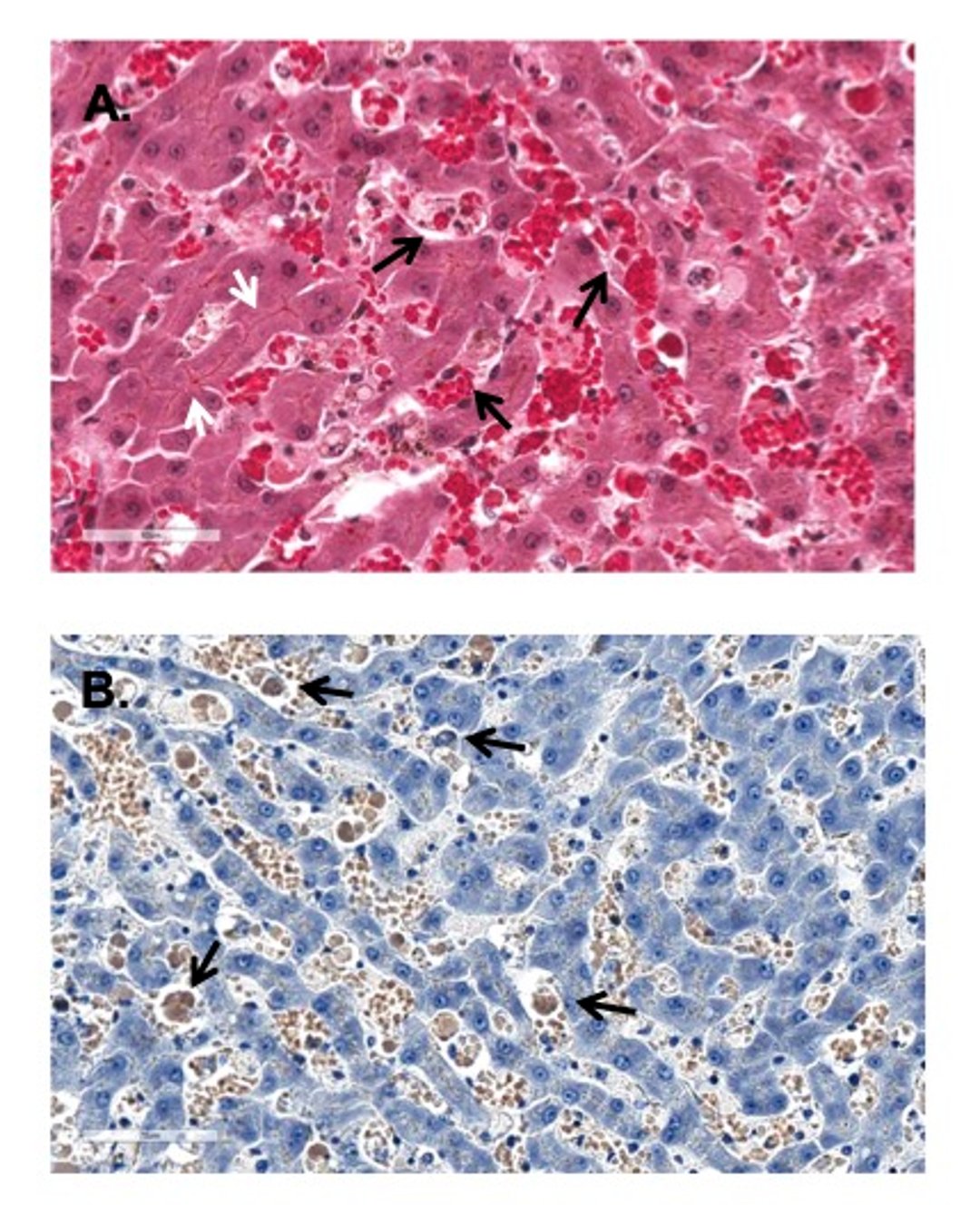
Photomicrograph of liver biopsies from a dog with agglutinating immune-mediated hemolytic anemia (IMHA) demonstrating severe obstruction of sinusoidal flow by agglutinated RBCs and Kupffer cells engorged with damaged phagocytized RBCs. A: Note agglutinated RBCs (black arrows) and occasional Kupffer cells filled with phagocytized RBCs. Extreme hyperbilirubinemia due to hemolysis has caused canalicular cholestasis (white arrows). Globular free hemoglobin is observed in sinusoids (cell free red globs). H&E stain. B: Liver section from the same dog. The tinctorial characteristics assist with visualizing agglutinated RBCs and Kupffer cells engorged with phagocytized RBCs (black arrow). Rhodanine stain; scale bar = xxx mcm.
Courtesy of Dr. Sharon Center.
A similar sinusoidal impact may develop secondary to Kupffer cell activation and scavenging of sinusoidal debris (eg, cellular debris from dead cells or inflammation; material transcending the portal circulation).

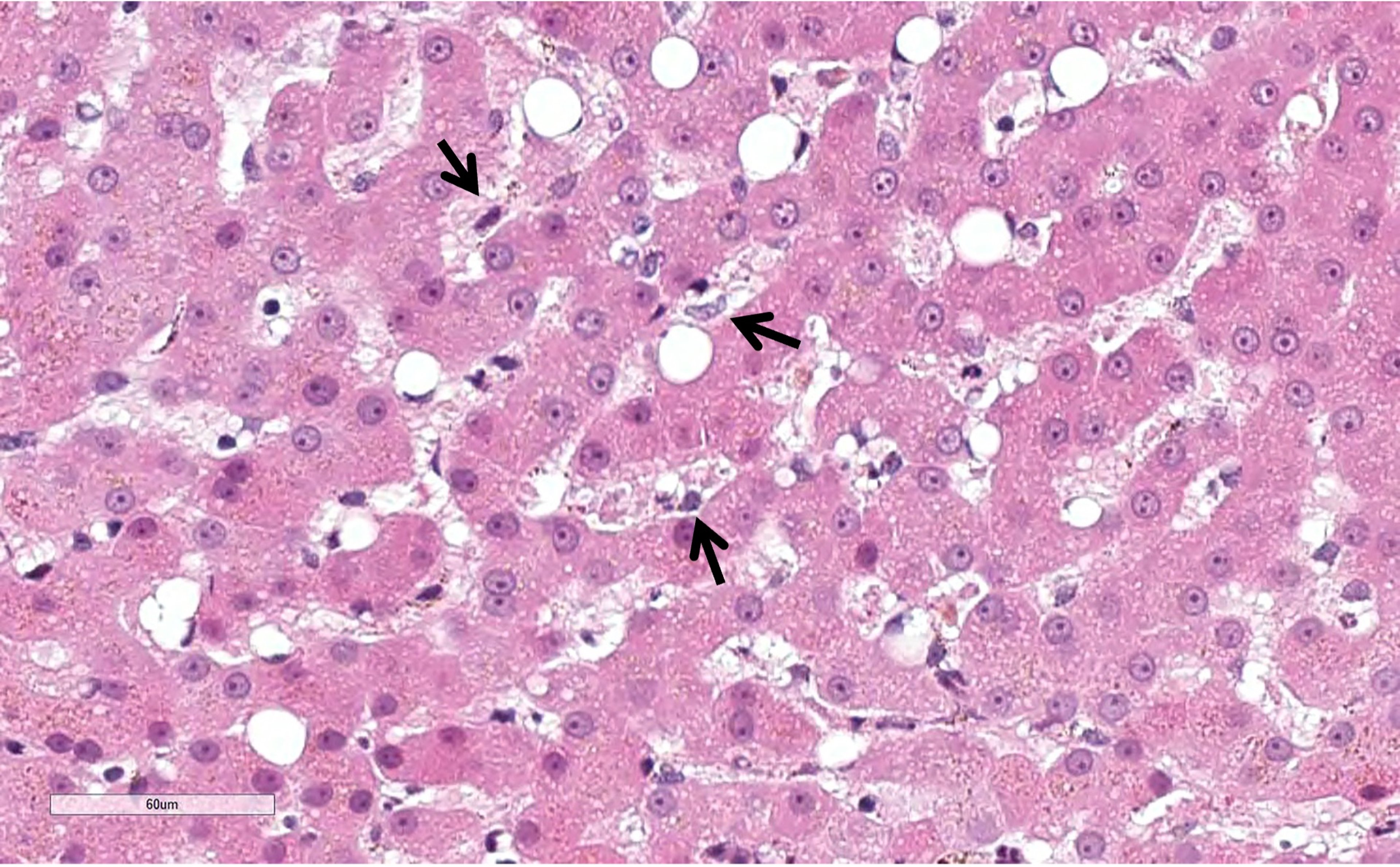
Photomicrograph of liver biopsy from a dog demonstrates Kupffer cell activation, hypertrophy, and marked phagocytosis of sinusoidal fluxing cellular and particulate debris. Enlarged Kupffer cell profiles (black arrows) and cellular debris partially compromise transhepatic perfusion. H&E stain; scale bar = 60 mcm.
Courtesy of Dr. Sharon Center.
The impact of compromised sinusoidal perfusion is complicated by the extent of transhepatic circulatory disruption, severity of the causal disorder, antecedent liver injury, and presence of hepatic functional insufficiency. Disrupted sinusoidal perfusion may notably compromise hepatocyte bilirubin elimination secondary to centrilobular hypoxia (centrilobular regions predisposed to hypoxia as the last lobular region receiving oxygenated blood).
Nonregenerative anemia may reflect anemia of chronic disease in some animals.
Regenerative anemia may reflect several complications, such as notable or occult enteric bleeding. Overt bleeding is characterized by melena, hematochezia, or both, or it may be insidious.
Enteric hemorrhage in advanced-stage liver disease or other disorders causing splanchnic hypertension is most often first encountered during the interval of decompressive compensation (ie, when acquired portosystemic shunts are developing ~4–6 weeks from onset of splanchnic hypertension). This phenomenon resembles hypertensive vasculopathy characterized in humans. The site of insidious hemorrhage in this scenario is often near the pyloro-duodenal junction.
Rarely, acquired decompressive portosystemic shunts cause severe hematuria. This occurs when shunting vasculature anastomoses with the wall of a ureter (or more rarely the kidney or urinary bladder).
Regenerative anemia may be encountered in animals with complete extrahepatic bile duct obstruction (EHBDO) or small- to medium-sized bile duct ductopenia. The regenerative anemia in this scenario reflects deficiency of vitamin K1 due to its malabsorption in the absence of enteric bile acids. Such animals are always jaundiced, usually for at least 7–10 days.
Administration of vitamin K1 (0.5–1.5 mg/kg) with three doses administered at 12-hour intervals typically curtails bleeding tendencies in these disorders. However, this is only a temporary solution. Preemptive intramuscular vitamin K administration should be provided as a routine preoperative mandate for jaundiced animals (without evidence of hemolysis) undergoing surgical interventions.
Poikilocytosis
Altered RBC morphology associated with liver disease is exhibited in cats more often than in dogs.
Cats with severe hepatic lipidosis (HL), cats with severe chronic lymphocytic or suppurative cholangiohepatitis, and cats with chronic (> 3 weeks EHBDO) may develop poikilocytosis (irregularly irregular RBC profiles) in the absence of schistocytosis.

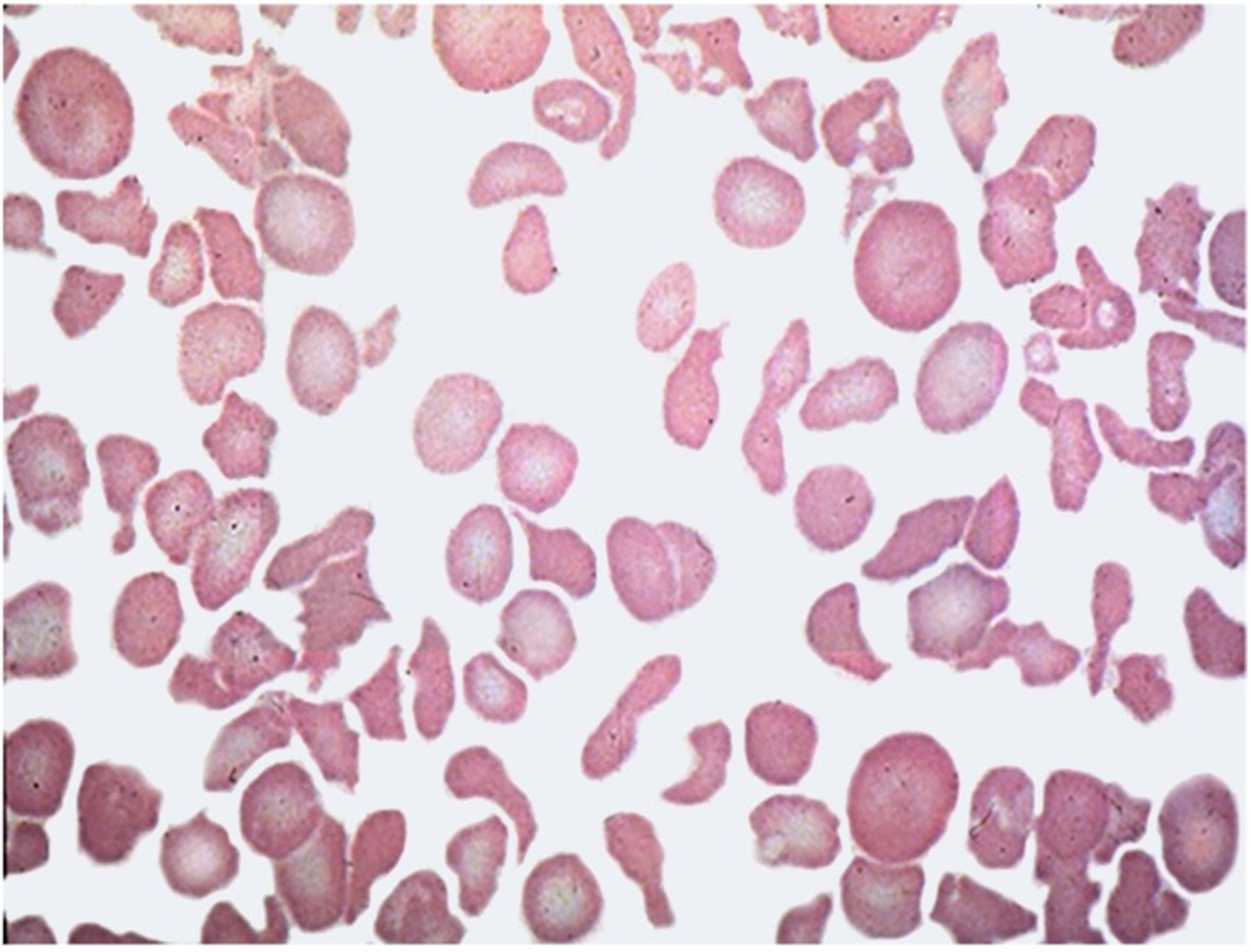
Photomicrograph of a blood smear from a cat with nonsuppurative cholangiohepatitis in which poikilocytosis is evident. Wright-Giemsa stain; original magnification, 1,000×.
Courtesy of Dr. Sharon Center.
The underlying cause of poikilocytosis is not known, although it has been conjectured to involve altered membrane integrity or structural modification of RBC membrane phospholipids. It is not associated with hypocobalaminemia (low B12 status). It remains possible that poikilocytosis might reflect sinusoidal compression in cats with HL where sinusoids are microscopically indistinguishable (see ). Similarly, dense portocentric inflammatory infiltrates and portal tract fibrosis in cats with severe cholangiohepatitis may impose a similar perfusion barrier.

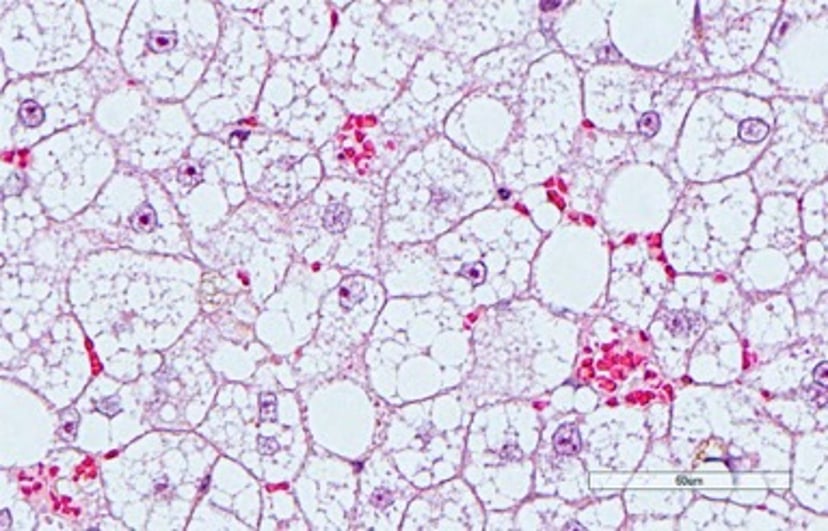
Photomicrograph of a liver section from a cat with severe microvesicular hepatic lipidosis demonstrating marked hepatocyte distention with triglyceride. Hepatocyte distention has compressed hepatic sinusoids and disturbs perfusion. Note congested RBC in scattered pockets, despite the fact that all hepatocytes abut sinusoidal vasculature. This phenomenon may contribute to transaminase release (ie, ALT and AST) from hepatocytes in cats with severe hepatic lipidosis. H&E stain; scale bar = 60 mcm.
Courtesy of Dr. Sharon Center.
Heinz Bodies
Cats with severe HL or CCHS may exhibit Heinz bodies, with or without anemia.
It is well established that cats are at higher risk, compared to dogs, for development of circulating Heinz bodies and associated hemolytic anemia. Circulating Heinz bodies are commonly encountered in cats with ketoacidotic diabetes mellitus, hyperthyroidism, and systemic lymphoma, as well as many other chronic illnesses with an oxidative injury mechanism.
This propensity reflects the unique sensitivity of feline hemoglobin to oxidative injury because of especially vulnerable hemoglobin sulfhydryl (-SH) groups. An additional factor is the nonsinusoidal splenic circulation in cats that inefficiently “pits” Heinz bodies from RBC membranes.
are microscopically identified “clumps” of oxidized (denatured) globin appearing as nipple-like protrusions at the surface of the red cell margin.

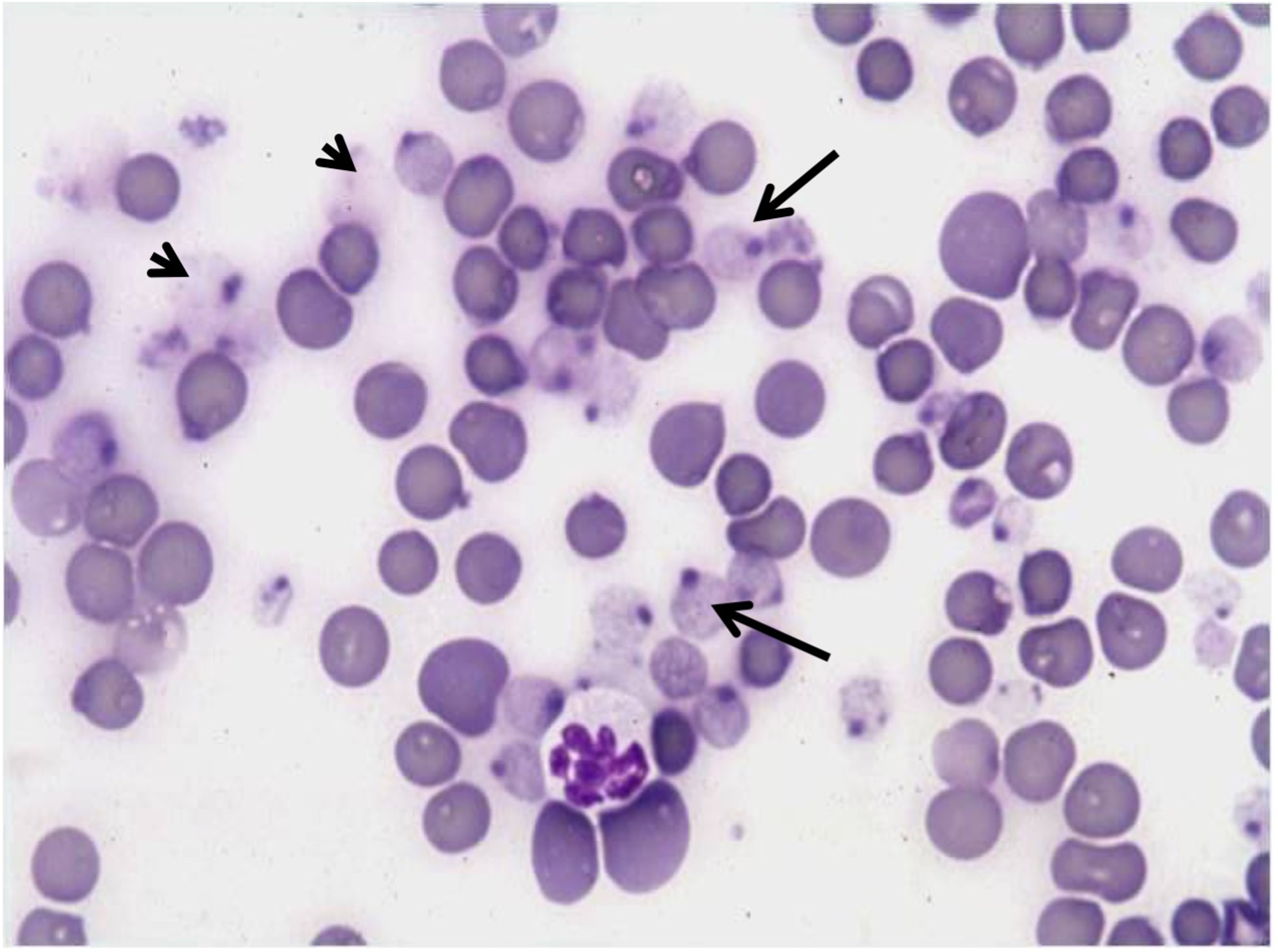
Photomicrograph of a blood smear from a cat with severe hepatic lipidosis demonstrating marked Heinz body hemolytic anemia. Lightly stained RBCs (arrows) are devitalized and hemolyzing with notable dark staining denatured globin (Heinz body) on the cell surface. A few “ghost” cells are present (remnant membranes of hemolyzed RBC, arrowheads). A single hypersegmented neutrophil is shown. New methylene blue stain distinguishes Heinz bodies. Counterstained with a modified Wright-Giemsa stain; original magnification, 1,000×.
Courtesy of Dr. Sharon Center.
Pioneering studies of feline Heinz bodies identified subnormal RBC concentrations of reduced glutathione (GSH; the form of GSH that provides antioxidant protection, oxidized GSH = GSSG) significantly lower than those in normal cats lacking RBC Heinz bodies. This study predicted what has been shown to be a cause and effect relationship: the subnormal concentrations of reduced GSH in RBC coordinated with a subnormal ATP concentration. Furthermore, RBCs from affected cats exhibited significantly decreased cell deformability, essential for navigating microvasculature, such as hepatic sinusoids.
Cats jaundiced secondary to severe HL, severe CCHS (nonsuppurative or suppurative), or chronic EHBDO exhibit risk for Heinz body–associated hemolytic anemia. Liver tissue from cats with these disorders similarly display subnormal reduced-GSH concentrations relative to normal cats. Thus, the microenvironment of oxidative liver injury is collaterally burdened by perfusion by RBCs with less flexible membranes, vulnerable hemoglobin -SH groups, and subnormal reduced-GSH concentrations.
The author has witnessed numerous cats with these disorders (at risk for Heinz body–associated hemolysis) develop Heinz body hemolysis ~8–12 hours after use of propofol for induction of anesthesia for esophageal feeding tube insertion. Notably, the hemolysis was often associated with clinical signs in the middle of the night. The severity of hemolysis caused a ~10%–18% decline in PCV and in some cases necessitated a blood transfusion. Risk for hemolysis with propofol has been previously noted in experimental studies in rodents, human infants, and cats—but not to the severity witnessed in these animals.
Other causes of Heinz body hemolytic anemia in cats (with and without liver disease) should always be considered. Common alternative causes include consumption of onions in food and dietary intake or drug vehicle exposure to propylene glycol (1,2-propanediol; can be a carrier for some IV administered drugs such as etomidate and diazepam).
Additionally, critical Heinz body hemolysis and anemia, requiring blood transfusion, have been witnessed in cats with liver disease that have been given repeated vitamin K1 injections (at a dosage of 5 mg/cat, IM, every 24 hours for > 2 weeks). Methemoglobinemia is often a first clinical feature of acetaminophen toxicosis in cats and dogs. Thereafter, hepatic necrosis develops if the animal survives for several days. Intravenous infusion of N-acetylcysteine is the first-line antidote using a protocol described in the section on feline hepatic lipidosis.
There are many other causes of Heinz body anemia in cats and dogs.
Critical Hypophosphatemia
Severe hypophosphatemia may develop secondary to a refeeding syndrome that leads to critical hypophosphatemia and hemolysis severe enough to warrant blood transfusion. This is encountered in catabolic anorectic cats with severe HL or CCHS.
The term “refeeding syndrome” was originally coined to describe a lethal syndrome witnessed in humans subjected to concentration camp food deprivation at the time of rescue and refeeding.
This lethal metabolic complication can be avoided by provision of IV fluid therapy judiciously supplemented with potassium phosphate and potassium chloride. Supplements are started concurrent with the start of nutritional support and carried through the first 3–7 days as needed.
In nontreated cats, hemolytic anemia develops ~24–48 hours after onset of extreme hypophosphatemia (< 2.0 mEq/L; see feline hepatic lipidosis).
Erythrocytes generate ATP from glycolysis, an enzymatic cascade regulated at several steps by inorganic phosphate. Consequently, extreme hypophosphatemia can limit glycolysis and ATP generation, which is the sole source of energy in these cells. Energy depletion causes RBC membrane rigidity (spherocyte or echinocyte shapes) with subsequent membrane damage during microvascular transit (ie, hepatic sinusoids).
The exact pathogenesis of membrane rigidity remains unclear aside from its link with ATP depletion. Proposed hypotheses include the following:
interrupted Na-K cell membrane pump secondary to ATP depletion
depletion of membrane-associated actin and myosin fibrils, known to influence cell membrane plasticity
altered composition of cell membrane phospholipids, compromising membrane flexibility
Refeeding syndrome also provokes critical clinical hypokalemia (weakness, gastric atony, other features). Preemptive administration of IV potassium phosphate, judiciously adjusted with potassium chloride (to meet but not exceed estimated potassium requirements) is used to avoid and manage this potentially lethal metabolic complication in at-risk cats. (See hepatic lipidosis.)
Increased risk for clinical hypophosphatemia also exists for cats with ketoacidotic diabetes mellitus because of their catabolic metabolism, electrolyte flux provoked by insulin administration, and disrupted glucose metabolism. Notably, a subset of cats with HL and CCHS also have diabetes mellitus, with escalated risk for refeeding syndrome.
Microcytosis
Development of erythrocytic microcytosis is associated with portosystemic shunting in dogs and cats. The cause of this phenomenon has not been determined despite investigation of circulating iron indices, bone marrow iron sequestration, measurement of circulating ferritin concentrations, examination of liver for iron sequestration, and assessment of hepcidin gene expression. Hepcidin, a peptide hormone synthesized in the liver, is the “master” regulator of systemic iron homeostasis.
High hepcidin expression suppresses enteric iron uptake and macrophage iron recycling, causing iron-restricted erythropoiesis (could be microcytic). Normal hepcidin expression has been shown in dogs with congenital portosystemic shunts.
Microcytosis is one of the most common clinicopathologic abnormalities associated with hepatofugal portal circulation (blood flow away from or around the liver as with portosystemic shunting) in dogs and cats. This hematologic abnormality is best interpreted in regards to the potential for portosystemic shunting when considered along with the following diagnostic features:
paired premeal and 2-hour postprandial bile acid concentrations (food withholding is not necessary for this test, but paired values must be measured on the same day; often both interval values are markedly increased in animals with portosystemic shunting of any cause)
total cholesterol concentration (hypocholesterolemia is common in shunting; dogs < 150 mg/dL, cats < 100 mg/dL)
subnormal BUN and creatinine concentrations
ammonium biurate crystalluria
with severe shunting, subnormal protein C activity (< 70%). Protein C is an anticoagulant serine protease (note that protein C is not related to and should not be confused with C-reactive protein). While protein C is helpful in distinguishing dogs with serious portosystemic shunting in this context, it has not been found to be useful in cats.
Microcytosis is also caused by iron deficiency. This should be ruled out as a causal factor on the basis of the patient’s history, physical examination, and other diagnostic assessments.
Up to 35% of cats with clinical hyperthyroidism also may exhibit microcytosis that self-resolves with effective management of the endocrinopathy.
In dogs, microcytosis is also a breed-associated peculiarity in Akitas, Chinese Shar-Peis, and Shiba Inus.
Schistocytes and Acanthocytes
Dogs with diffuse severe necroinflammatory liver disease, fulminant hepatic failure with lobular collapse, chronic sinusoidal fibrosis, or advanced-stage liver disease (previously referred to as cirrhosis) have compromised sinusoidal perfusion. In some of these diseases, irregular, narrow, and turbulent sinusoidal rheology leads to microvascular RBC shearing, generating schistocytes and acanthocytes. This morphological pattern is not inexorably linked with disseminated intravascular coagulation (DIC).
Leukocytes
Leukograms in animals with hepatobiliary disease are widely variable, inconsistent, and nonspecific because of the breadth of causal factors, severity of liver disease, presence or absence of a necroinflammatory phenotype, and shared systemic disease.
Leukocytosis may reflect inflammatory, infectious, endotoxic, necrolytic, or diffuse infiltrative hepatic disorders, release of endogenous glucocorticoids, or glucocorticoid administration.
Leukopenia can reflect sepsis or toxicosis that either provokes development of a sump where cells accumulate in the liver or may reflect a shared injury involving the bone marrow.
Thrombocytopenia
In animals with severe diffuse necroinflammatory hepatic parenchymal or sinusoidal injury, damaged microvasculature can provoke platelet aggregation and sequestration, contributing to thrombocytopenia with or without syndromic features of disseminated intravascular coagulation (DIC). Thrombocytopenia also reflects decreased hepatic thrombopoietin production and vascular mediated injury by infectious agents or immune mechanisms.
Thrombocytosis in humans with portal hypertension was long considered to reflect splenic sequestration; however, it is now thought to be importantly influenced by decreased thrombopoietin expression due to decreased functional hepatic mass.





