




Chronic wasting disease is a transmissible spongiform encephalopathy that affects deer and other cervids, primarily in North America. It is a fatal, progressive neurodegenerative disorder and affects both wild and farmed animals. The primary signs are significant weight loss, ataxia, and hypersalivation. Diagnosis is made by ELISA and Western blot, with confirmation by immunohistochemistry. There are no treatments or vaccines, so control in farmed animals relies on depopulation of affected herds.
Chronic wasting disease (CWD) is a contagious and fatal neurodegenerative disease of captive and free-ranging cervids, including deer, elk, moose, and reindeer. It is a member of the transmissible spongiform encephalopathy (TSE) family of diseases, or prion diseases, that includes bovine spongiform encephalopathy; scrapie of sheep and goats; transmissible mink encephalopathy; and kuru, Creutzfeldt-Jakob disease (CJD), and variant CJD of humans.
All of these diseases are caused by prions, infectious agents consisting solely of protein that do not use encoding nucleic acid for replication. They consist of PrPSc, a misfolded and aggregation-prone isoform of the host-encoded cellular prion protein (PrPc). Upon direct binding of PrPc to PrPSc, PrPc adopts the disease-associated conformation that is the basis for prion replication. In contrast to PrPc, PrPSc is partially resistant to protease digestion and accumulates within neurons, which eventually leads to neuronal death.
The main symptoms of CWD, which occur after a long incubation period up to several years, are significant weight loss, ataxia, and hypersalivation. Affected animals are separated from the herd. There is no curative or prophylactic treatment available.

Courtesy of USGS and Bryan Richards, USGS National Wildlife Health Center. Public domain.
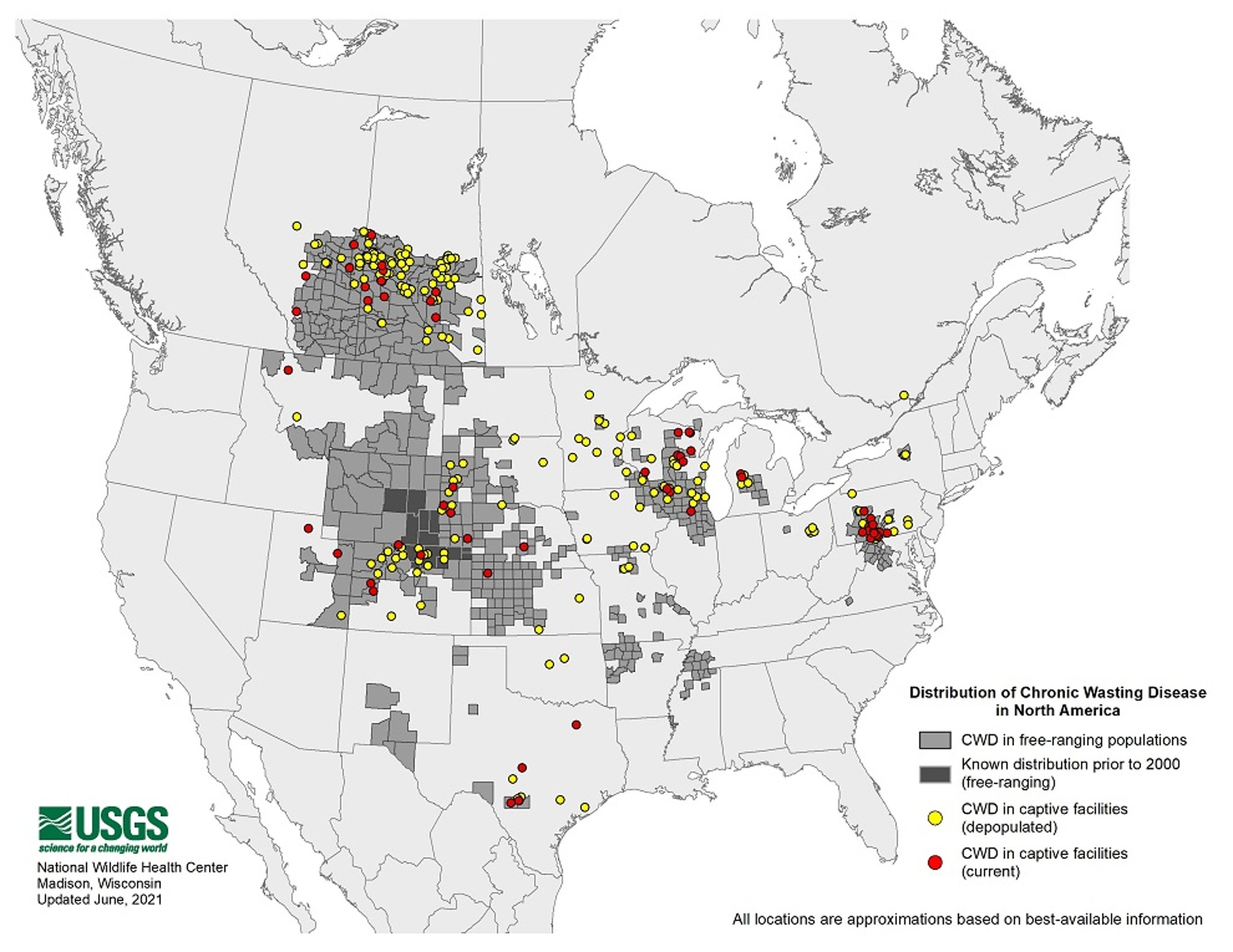
CWD was first identified as a clinical syndrome in the late 1960s among captive mule deer in Colorado; a decade later, it was recognized to be a spongiform encephalopathy with characteristics similar to those of scrapie . It is found in farmed or free-ranging populations of mule deer, white-tailed deer, red deer, and elk (wapiti) in 26 states (USA) and 3 Canadian provinces, with the most recent identification of CWD in captive red deer in Quebec, Canada. Rare cases of CWD in free-ranging moose have been diagnosed in Colorado and in Alberta, Canada. Many states and provinces have developed regulations for control and management of CWD in farmed populations, and federal regulations are in place in Canada and the USA. It is a reportable disease in most jurisdictions.
CWD has been identified outside of North America in South Korea, where a few elk imported from Canada were CWD infected. In 2016, CWD was diagnosed in wild reindeer in Norway, which marked the first finding of the disease in Europe and the first case in wild reindeer. Since then, CWD was discovered in several Norwegian red deer and moose, and in 2018 and 2019, single cases in moose were documented in Finland and Sweden.
Etiology of Chronic Wasting Disease
Chronic wasting disease is mostly acquired by infection with CWD prions. It is known to naturally affect mule deer, white-tailed deer, elk, red deer, and moose. Experimentally, CWD can be transmitted by intracerebral inoculation to cattle, sheep, goats, domestic ferrets, mink, mice, hamsters, and squirrel monkeys. Transmission efficiency to cattle varies depending on the cervid species from which the CWD-infected brain homogenate used for infection is derived.
A large study to investigate susceptibility of cattle by the more natural route of oral or contact exposure began in 1997, and there has been evidence of CWD in the experimentally exposed cattle. Differences in species susceptibility to CWD probably relate to sequence differences among normal host PrP proteins.
Investigations of the recent cases in moose and red deer in Scandinavia suggest an additional form of atypical CWD. In these animals, PrPSc distribution was restricted to the brain, and affected animals were approximately 15 years old; therefore, a sporadic origin of the disease in these cases is possible.
Transmission, Epidemiology, and Pathogenesis of Chronic Wasting Disease
Chronic wasting disease is transmitted horizontally and vertically. Because prions are highly resistant to environmental and chemical inactivation, they may accumulate in the environment and thus be available to infect susceptible cervids. Consequently, close confinement of farm-raised cervids will likely potentiate the spread of CWD. Likewise, winter feeding of deer and elk will concentrate cervid populations and likely potentiate horizontal transmission in wild populations. Foraging on feeding grounds contaminated by urine or feces of infected animals or contact with either infected animals or decomposing carcasses of animals with CWD results in transmission of disease to other susceptible cervids.
The agent probably enters a susceptible host via ingestion and is taken up by lymphoid tissues associated with the GI tract. The agent can be detected in lymphoid, nervous tissues, muscle tissue, antler velvet, blood, saliva, urine, and feces. Prions can be detected in the blood and saliva of infected animals as soon as 3 months after infection. In urine and feces, detection of prions is also possible already at a preclinical stage of the disease.
The agent most likely arrives in the brain by retrograde movement up the vagus nerve to the dorsal motor nucleus of the vagus at the obex region of the medulla oblongata. Spongiform lesions in the brain develop first in the vagal nucleus at about the time of onset of clinical disease. This occurs naturally with an incubation period of ~1.5–3 years; however, incubation periods in all species known to be affected by CWD are influenced by certain PrP polymorphisms. Prevalence in captive herds of deer and elk may reach nearly 100% in heavily contaminated facilities; prevalence in free-ranging cervids is extremely variable, from < 1% to 40%, in some hunting areas in Wyoming.
The movement of CWD in populations of free-ranging deer and elk follows natural migration routes, often along waterways and natural corridors. In the past, movement of CWD in farmed deer and elk in commerce was through human-facilitated transportation of animals incubating CWD. Now that programs and regulations are in place in most jurisdictions, movement of CWD in live animals should be curtailed. Surveillance, however, should continue, and submission of samples from hunter-harvested animals may be mandatory in areas with a high prevalence of CWD.
Clinical Findings of Chronic Wasting Disease
Animals with clinical chronic wasting disease are >16 months old and show a spectrum of signs. The earliest and most difficult to appreciate are subtle changes in behavior and weight loss. These changes are often detectable only by animal caretakers familiar with the individual animal. As the disease progresses, behavioral changes may include alterations in how the animal interacts with herdmates and caretakers, loss of wariness, somnolence, persistent walking, polydipsia and polyuria, and hyperexcitability when handled. Affected animals may show variable locomotor signs, including ataxia (especially posterior ataxia) and head tremors.
Late in the disease, animals may have a low head carriage, drooped ears, and fixed staring gaze; they may hypersalivate and grind their teeth. Death after routine chemical immobilization has been noted. Aspiration pneumonia may be the only presenting clinical sign and is often the cause of death. CWD should be suspected in any adult cervid with aspiration pneumonia. Weight loss is progressive throughout the course of disease, even when adequate feed is present, but it is important to recognize that CWD may be present in cervids that are not emaciated. Death of CWD-affected animals may be precipitated by cold weather or other acute stressors. Affected cervids are more susceptible to hunting, predation, vehicle collisions, and other forms of death by misadventure. Carcasses and offal should be disposed of in a manner that limits the exposure of farm-raised and free-ranging cervids to any potentially infectious material.
Lesions
Lesions from chronic wasting disease are seen in the gray matter of the CNS. Lesions are bilaterally symmetrical and anatomically constant among animals. Spongiform appearance is obvious; vacuolization occurs in neuronal perikarya and neuronal processes. Along with neuronal degeneration, astrocytic hyperplasia and hypertrophy may appear. H&E staining of brains of affected animals reveals amyloid plaques that appear as pale, fibrillar, eosinophilic areas of neuropil and are sometimes surrounded by vacuoles (florid plaques).
Detection of PrPSc in brain sections by immunohistochemistry (IHC) provides a very good way to visualize CWD pathology while maintaining the structural context. PrPSc is detected by IHC or immunoblot in a variety of tissues in cervids showing clinical signs. Although PrPSc can be found in regions of the brain not exhibiting spongiform change, typically there is correlation between PrPSc deposition and spongiform appearance.
Diagnosis of Chronic Wasting Disease
Diagnosis is based on detection of PrPSc by ELISA or Western blot, with confirmation by immunohistochemistry
Diagnosis of chronic wasting disease based on clinical signs is not reliable, because they are unspecific and mild at the beginning of disease. Therefore, it relies on the detection of protease-resistant PrPSc by ELISA or Western blot and confirmed by immunohistochemical detection of PrPSc in brains (obex region) or lymphatic tissue.
In mule deer and white-tailed deer, the CWD PrP accumulates in the retropharyngeal lymph node before arriving in the brain; thus, it is considered to be the most important tissue to collect for testing. Both brain and lymph node samples should be collected from elk. The correct portion of the brain (ie, the obex, at the caudal end of the fourth ventricle below the cerebellum) must be collected for a meaningful test.
The laboratory should be consulted to determine whether the samples must be fixed in 10% buffered formalin, chilled or frozen, or if portions of the samples should be sent both fixed and frozen. Samples should be, and in many jurisdictions are required to be, submitted to certified laboratories to be tested for evidence of CWD. It is good practice either to send the carcass to the diagnostic laboratory or to collect a wide variety of samples, so that if diseases other than CWD are present they will be identified. At a minimum, samples for CWD testing should include brain and retropharyngeal lymph nodes. Many laboratories accept whole heads from cervids for testing.
Surveillance programs for free-ranging cervids vary depending on the jurisdiction and are usually conducted by the local wildlife management agency, which should be consulted if CWD is suspected in a free-ranging deer or elk. Surveillance depends mainly on submission of heads from hunter-harvested animals. In some areas, active surveillance is done by taking biopsies from tonsils, retropharyngeal lymph nodes, and recto-anal mucosa-associated lymphoid tissue (RAMALT). Diagnostic tests include detection of PrPSc by IHC, ELISA, or Western blot in brain and/or lymphoid tissues. In the USA, these tests are run only at USDA-certified laboratories. ELISA is used as a screening test, and IHC, which is considered the preferred test, is used to confirm positive ELISA.
Throughout the past decade, in vitro conversion methods for amplification and detection of minute amounts of PrPSc such as protein misfolding cyclic amplification or real-time quaking-induced conversion assay have been developed. Using these tests, CWD prions are detectable at a preclinical stage in specimens that can be obtained antemortem by noninvasive methods, such as blood, urine, feces, saliva, or nasal brushings. These newly developed assays are undergoing validation.
Differential diagnoses for animals suspected of CWD include:
brain abscesses
traumatic injuries
meningitis
encephalitis
peritonitis
pneumonia
arthritis
starvation and nutritional deficiencies
dental attrition
anesthetic death
Treatment and Control of Chronic Wasting Disease
There are no treatments, prophylaxis, or supportive care available for any TSE. Control in farmed cervids is by depopulation with indemnity and development of CWD herd certification programs in the USA and Canada.
These are voluntary cooperative programs between industry and federal or state/provincial governments. They are administered by the USDA Animal and Plant Health Inspection Service and the Canadian Food Inspection Agency. This plans typically require 5 years of monitoring to achieve the highest status. The bases for CWD control programs in the farmed cervid industry are individual animal identification, CWD testing in all animals in the herd that die over a certain age, and limiting new herd additions to animals from herds of comparable or higher CWD status.
Control of chronic wasting disease in free-ranging populations is extremely difficult. All jurisdictions have banned movement of live cervids from endemic areas for translocations, and many have regulations on movement of parts of hunted deer and elk. In areas where CWD occurs, attempts at control have included population reduction, test and removal, and intensified surveillance, but these have had limited success.
Only a few disinfectants and methods of disposal inactivate prions. Fresh household bleach at 50% concentration for 30–60 minutes or sodium hydroxide (1 M) for 60 minutes will inactivate the agent. This is inexpensive and readily available but may be corrosive to some surfaces and instruments. Additional disinfectants are being considered for general use but are not yet approved. Incineration in a medical incinerator, alkaline digestion in specially designed equipment, and disposal in certified municipal landfills are used for tissues and carcasses of animals with CWD.
Zoonotic Risk of Chronic Wasting Disease
Although chronic wasting disease has been present in hunted populations of deer and elk for >30 years, no case of human CWD has been identified. The risk to people appears to be minimal. The CWD agent has been detected in the muscle tissue of infected cervids. Public health authorities and wildlife management agencies suggest the following precautions for hunters and people handling cervids in areas where CWD is found to further reduce risk of human exposure:
do not harvest deer or elk that appear to be sick or abnormal
wear rubber, plastic, or latex gloves when dressing the carcass
avoid contact with brain, spinal cord, and lymphoid tissues
debone the meat when processing
disinfect knives, saws, and tables with 50% bleach
have the animal tested for CWD
All public health authorities recommend that animals positive for any TSE not be consumed by people or other animals.
Key Points
Chronic wasting disease is a fatal and transmissible neurodegenerative disease of wild and captive cervids caused by prions.
It has been reported in 26 US states, 3 Canadian provinces, and 3 European countries.
Clinical signs include progressive weight loss, ataxia, hypersalivation, and behavioral changes.
CWD is diagnosed mostly postmortem by detection of disease-associated PrPSc in brain or lymphatic tissue.
No therapy or prophylaxis is available.




