




The goal of drug treatment is to achieve a pharmacological response. According to drug-receptor theory, the magnitude of the pharmacodynamic response to a drug generally reflects the number of receptors with which the drug interacts. Because tissue drug concentrations generally parallel plasma concentrations, the ideal dose will result in plasma drug concentrations in a therapeutic range. This population statistic is defined by a maximum drug concentration, above which the risk of adverse events (eg, toxicosis) increases, and a minimum drug concentration, below which therapeutic failure may result.
Pharmacokinetics is the branch of pharmacology concerned with mathematical description of the time course of plasma drug concentrations measured after administration of a dose. Plasma drug concentrations generally fluctuate during a dosing interval, being affected by the simultaneous action of the four basic drug movements: absorption, distribution, metabolism, and excretion (together known as ADME). After absorption from the site of non-IV administration into the plasma, and distribution into tissues and then back into plasma, the drug can then be eliminated from the body by either metabolism or excretion of the parent drug or its metabolites.
Many factors influence each of these four drug movements and thus also the time course of plasma drug concentrations after a dose is administered by any route. Understanding these factors, in turn, is important for individualizing drug treatment for the patient, because dosing regimens are modified to adjust for physiologic (eg, species, breed, sex and neuter status, age), pharmacological (eg, drug-drug or drug-diet interactions), and pathological (eg, renal, hepatic, or cardiac disease) influences on drug disposition.
Passage of Drugs Across Cell Membranes
Each of the four drug movements (absorption, distribution, metabolism, and excretion) generally relies on the drug passing through cell membranes (transcellular movement). Membrane barriers may be composed of several layers of cells (eg, skin, vagina, cornea, placenta) or a single layer of cells (eg, enterocytes, renal tubular epithelial cells), or they may consist only of a boundary less than one cell in thickness (eg, hepatic sinusoids). Multilayered tissues each may present different types of barriers (eg, skin is protected by the dense stratum corneum, which is absent in mucous membranes).
Not all drugs must pass through cell membranes. Paracellular movement between cells is an important movement for some drugs (eg, in the GI tract).
Drugs and other molecules cross cell membranes by several processes. Methods by which drugs move include bulk flow (eg, movement with blood, glomerular filtration), passive diffusion, carrier-mediated transport (ie, active or facilitated transport), and pinocytosis. Of these, passive diffusion is the most important for the movement of drug molecules and other xenobiotics (substances foreign to the body), as well as many endogenous compounds.
The rate at which a drug passively diffuses through membranes is influenced by several factors, the most important of which is the concentration gradient of diffusible (eg, dissolved) drug across the membrane. However, other host and drug factors influence the rate and extent of passive diffusion.
Host factors that increase diffusion include permeability and surface area of the membrane. Thickness of the membrane negatively affects diffusion. Drug characteristics that influence diffusion include molecular weight, lipophilia, and extent of ionization.
Most drugs are small molecules (< 900 daltons). Diffusibility is greatest for drugs with low molecular weight (< 500 daltons).
Drugs must be sufficiently lipid soluble (lipophilic) to pass through some level of cell membrane lipid bilayer to reach most drug receptors. The lipid-water partition coefficient describes the distribution (ratio) of a drug (concentration) in a lipid compared with water media. The distribution coefficient also takes into account ionization.
Many drugs are weak organic acids or bases. At physiologic pH, they tend to be partially ionized (dissociated) and partially nonionized (undissociated). The ratio of the respective forms depends on the dissociation constant (pKa) of the drug—ie, the pH at which the drug is present in equal concentration in ionized and nonionized forms—and the pH of the solution in which the drug is dissolved. Only the nonionized fraction diffuses through lipid membranes. Distribution across any membrane of a drug with any given pKa reflects the extent of ionization and thus environmental pH on each side of the membrane.
The Henderson-Hasselbalch equation predicts the ratio of ionized versus nonionized drug. In general, weak acids are nonionized in acidic compared with alkaline environments, and weak bases are nonionized in alkaline compared with acidic environments. The more similar the environmental pH is to that of the pKa of a weak acid or base, the more nonionized the drug is and the more likely it is to diffuse. As long as the ratio of nonionized to ionized drug is ≥0.01, the drug is considered diffusible.
Not all drugs must pass through cell membranes to reach their receptor. Aqueous pores in lipoproteinaceous biological membranes offer a means of xenobiotic movement through the membrane for predominantly aqueous soluble drugs. Lipid-insoluble (water-soluble) compounds pass easily through these pores and, to a lesser extent, directly through the membrane. A difference in hydrostatic or osmotic pressure across a membrane facilitates movement by promoting water flow through the aqueous pores. Bulk fluid movement carries solute molecules through the pores, as long as the solute molecules are smaller than the aqueous channels.
Several specialized transfer processes account for the passage of certain organic ions and other large lipid-insoluble substances across biological membranes. Active transport, facilitated diffusion, and exchange diffusion are three distinct types of carrier-mediated systems used to move specific substances across cell membranes.
The highly selective carrier-mediated systems are used principally for transporting nutrients and natural substrates across biological membranes. Among the mechanisms of active transport are transport proteins that move compounds, including drugs, into or out of cells. Transport proteins are located at portals of entry (eg, enterocytes of the GI tract or sinusoidal hepatic cells) or sanctuary tissues (eg, brain, CSF, placenta, prostate, eyes, or testicles), where they help ensure that xenobiotics do not enter the protected tissue. Therefore, transport proteins are able to influence each drug movement (absorption, distribution, metabolism, and excretion).
The best known of the transport proteins is the ATP-binding cassette superfamily of efflux transporters, which includes P-glycoprotein, the multidrug resistance protein. Substrates for P-glycoprotein include both xenobiotics and dietary components. Additional transport proteins carry cations, anions, or organic compounds. Competition for transport increases oral absorption or distribution of one of the competing molecules.
Pinocytosis is an important transport process in mammalian cells, particularly intestinal epithelial cells and renal tubular cells. Drugs that exist in solution as molecular aggregates that have large molecular masses themselves or that are bound to macromolecules may be transferred across membranes by pinocytosis.
Drug Absorption
Absorption of Drugs From the GI Tract in Animals
Before absorption, orally administered drugs must disintegrate and then dissolve; dissolution often is the rate-limiting factor in oral drug absorption. After an orally administered drug has dissolved, multiple factors influence its absorption.
Drugs may be formulated by manufacturers such that absorption is extended (occurring throughout the GI tract) or delayed. Often, release kinetics of these drugs limits the extrapolation of dosing regimens from one species to another.
Drug characteristics include molecular size, liphophilia, and drug pKa. Drug pKa is particularly important in the GI tract because environmental pH is markedly variable among the different regions or compartments.
Host factors (in addition to environmental pH) that affect oral absorption include epithelial permeability, GI motility, surface area (being greatest in the small intestine, which is the major site of drug absorption), transport and metabolizing proteins, and GI blood (which maintains the concentration gradient) and lymphatic flow.
Epithelial permeability is influenced by disease. In addition, in species in which colostrum absorption is important, epithelial permeability is much greater shortly after birth.
GI motility influences the mixing of luminal contents, which is necessary for dissolved drugs to come into contact with absorptive surfaces. Gastric motility determines gastric emptying, which in turn influences the rate of drug absorption.
Changes in GI blood flow have minimal impact on drug absorption. Although GI blood flow maintains the concentration gradient necessary for passive diffusion, it is rarely the rate-limiting factor.
Both efflux transport proteins (eg, P-glycoprotein) and drug-metabolizing enzymes located in the GI epithelium can markedly decrease drug absorption, contributing to a first-pass effect. These proteins are subject to clinically relevant drug interactions and are likely to differ based on physiological influences (eg, species, sex and neuter status, age).
A drug entering the portal circulation will be exposed to hepatocytes before reaching the systemic circulation. If a large proportion of the drug (> 75%) is removed or extracted as it passes through the liver, the oral bioavailability of the drug will be markedly decreased, also resulting in a first-pass effect. For such drugs, the oral dose is proportionately higher than the parenteral dose.
Food can markedly alter the oral absorption of drugs by either diluting or, more importantly, binding to it so that it is not absorbed.

Absorption of Drugs From Topical Administration in Animals
Drugs may be absorbed through the skin after topical application; however, the stratum corneum presents an effective barrier to the transdermal movement of most drugs into circulation. Therefore, many drugs are absorbed after paracellular rather than transcellular movement. Intact skin allows the passage of small lipophilic substances but efficiently delays the diffusion of water-soluble molecules in most cases. Lipid-insoluble drugs generally penetrate the skin slowly, compared with their rates of absorption through other body membranes.
Absorption of drugs through the skin may be enhanced by heat, moisture, or disruption of the stratum corneum. Occlusive dressings increase heat and moisture; transdermal patches also disrupt the stratum corneum. Smaller molecules are more conducive to transdermal drug delivery.
Various transdermal systems have been developed with the intent of systemic drug delivery. Certain solvents (eg, dimethyl sulfoxide [DMSO]) may facilitate the penetration of drugs through the skin. Damaged, inflamed, or hyperemic skin allows many drugs to penetrate the dermal barrier much more readily. For example, drugs administered transdermally in pluronic lecithin organogel or other gels presumably penetrate the stratum corneum because it is disrupted, becoming more permeable.
The same principles that govern absorption of drugs through the skin pertain to topical preparations applied on epithelial surfaces. Because the mucosal epithelium has no stratum corneum, drugs are absorbed more quickly there than they are through the skin. An advantage of the buccal mucosa for drug absorption is that the drug avoids first-pass metabolism.
Absorption of Drugs From Tracheobronchial Surfaces and Alveoli
Because volatile and gaseous anesthetics have relatively high lipid-water partition coefficients and generally are rather small molecules, they diffuse practically instantaneously into the blood in the alveolar capillaries. In contrast, for drugs administered as aerosols, particles containing drugs can be deposited on the mucosal surface of the bronchi or bronchioles, or even in the alveoli; the site depends on particle size and breathing rate and depth. Whether the drug is absorbed from these sites depends on the same general principles that govern the passage of drugs across cell membranes.
Absorption of Drugs From Injection Delivery Sites
After non-IV injection, diffusible drug molecules traverse the capillary wall by a combination of diffusion and filtration. Diffusion is the predominant mode of transfer for lipid-soluble molecules, small lipid-insoluble molecules, and ions. Because most capillaries are fenestrated, all drugs, whether lipid soluble or not, cross the capillary wall at rates that are extremely rapid compared with their rates across other body membranes. In fact, the movement of most drug molecules in various tissues is limited only by the rate of blood flow rather than by the capillary wall. However, endothelial cells of blood-tissue barriers (eg, blood-brain barrier, blood-CSF barrier, blood-retinal barrier, and blood-testis barrier have tight intercellular junctions, thus restricting the movement of drugs into pharmacological sanctuary tissues.
Aqueous solutions of drugs are usually absorbed from an IM injection site within 10–30 minutes, as long as blood flow is unimpaired. Faster or slower absorption is possible, depending on concentration and lipid solubility of the drug, vascularity of the site (including number of vessels and state of vasoconstriction), volume of injection, osmolality of the solution, and other pharmaceutical factors. Substances with molecular weights > 20,000 daltons are principally taken up into the lymphatics.
The absorption of drugs from subcutaneous tissues is influenced by the same factors that determine the rate of absorption from intramuscular sites. Some drugs are absorbed as rapidly from subcutaneous tissues as from muscle, although absorption from injection sites in subcutaneous fat is always markedly delayed.
Increasing the blood supply to the injection site by heat, massage, or exercise hastens the rate of dissemination and absorption.
The rate of absorption of an injected drug may be altered in a number of ways. The following prolong absorption: immobilization of the site, local cooling, a tourniquet, incorporation of a vasoconstrictor, an oil base, and implant pellets and insoluble depot preparations. Among these depot preparations are drugs that are converted to less-soluble esters, which must be released by esterases (eg, procaine and benzathine esters of penicillin or acetate esters of steroids) or less-soluble complexes (eg, protamine zinc insulin), or that are administered as insoluble microcrystalline suspensions (eg, methylprednisolone acetate).
Drug Bioavailability in Animals
Bioavailability is a measure of the extent to which a drug is absorbed into the body and thus is available to act on its intended target site. It is calculated by comparing the areas under the plasma concentration curve following non-IV (eg, oral, buccal, ocular, nasal, rectal, transdermal, SC, or sublingual) and IV administration, normalized for body weight and drug dose, expressed as a percentage:

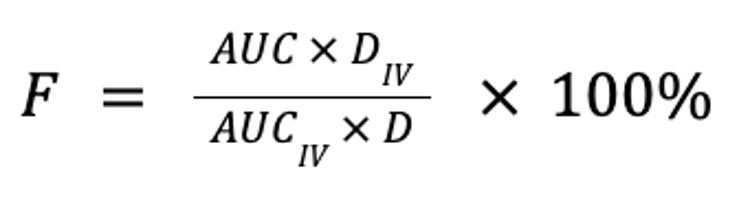
where F is the fraction of the drug that reaches the systemic circulation, AUC is the area under the plasma concentration curve, and D is the dose (in mg/kg). Note that for equivalent doses, bioavailability is simply the ratio of the respective AUC for the non-IV and IV routes:

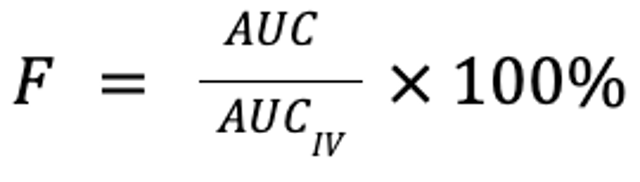
A drug is considered 100% bioavailable when administered IV (although pulmonary metabolism may make this not entirely true).
Two products are considered bioequivalent if they result in the same rate and extent of absorption. Rate of absorption is reflected as the maximum plasma drug concentration (Cmax) and the time to Cmax (Tmax). Thus, two drugs are bioequivalent if their AUC, Cmax, and Tmaxdo not differ substantially. Different preparations or routes of administration may be equally bioavailable but not bioequivalent. Generic drugs must be demonstrated as bioequivalent to their brand-name counterparts.
After a drug reaches the systemic circulation through absorption, all other drug movements (distribution, metabolism, and excretion) will be the same regardless of the route or preparation. In some instances, however, absorption may be so slow (eg, delayed or extended-release formulation, absorption affected by food intake) that it limits the rate at which the drug is eliminated (metabolized or excreted) from the body. The situation where the rate of absorption is slower than the rate of elimination is known as "flip-flop kinetics."
Drug Distribution
After being absorbed into the bloodstream, drugs are disseminated to all parts of the body. Occasionally, the drug molecule is so large (> 65,000 daltons) or so highly bound to plasma proteins that it remains in the intravascular space after IV administration. Compounds that pass freely through cell membranes become distributed, in time, throughout the body water to both extracellular and intracellular fluids, with the extent depending on drug chemistry. Substances that pass readily through and between capillary endothelial cells but do not penetrate other cell membranes are distributed into the extracellular fluid space.
Drugs may also be redistributed in the body after initial high concentrations are achieved in tissues that have a rich vascular supply—eg, the brain. As the plasma concentration falls, the drug readily diffuses back into the circulation to be quickly redistributed to other tissues with high blood-flow rates, such as the muscles; over time, the drug also becomes deposited in lipid-rich tissues with poor blood supplies, such as fat depots. Most drugs are not distributed equally throughout the body but tend to accumulate in certain specific tissues or fluids.
The general principles that govern the passage of drugs across cell membranes apply to drug distribution. Basic drugs tend to accumulate in tissues and fluids with pH lower than the pKa of the drug; conversely, acidic drugs concentrate in regions of higher pH, as long as the free drug is sufficiently lipid soluble to penetrate the membranes that separate the compartments. Even small differences in pH across boundary membranes, such as those that exist between plasma (pH, 7.4) and other compartments—eg, CSF (pH, 7.3), milk (pH, 6.5–6.8), renal tubular fluid (pH, 5–8), and inflamed tissue (pH, 6–7)—can lead to an unequal distribution of drugs, referred to as ion trapping.
Only freely diffusible and unbound drug molecules are generally able to pass from one compartment to another. However, some drugs are transported by carrier-mediated systems across certain cell membranes, leading to higher concentrations on one side compared to the other. Examples of such nonspecific transport mechanisms are found in renal tubular epithelial cells, hepatocytes, and the choroid plexus. Among the transport proteins, genetic differences in P-glycoprotein profoundly affect drug movement, particularly at portals of xenobiotic entry or sanctuary tissues.
Passive diffusion of drugs from capillaries to tissues may be limited by binding of the drugs to plasma proteins. The most important plasma protein is albumin; however, the globulins and, especially, alpha-1 acid glycoprotein (for bases) may also play an important role. In general, ≥ 80% protein binding is considered clinically relevant. For such drugs, factors that influence binding can affect drug disposition. These factors include plasma pH, concentration of plasma proteins, concentration of the drug, or the presence of a competing agent with a greater affinity for the limited number of binding sites. For example, a potentially toxic compound (eg, most NSAIDs) may be 98% bound, but if it becomes only 96% bound, then the concentration of the free active drug that becomes available in the plasma is doubled, with potentially harmful consequences.
The concentration of a drug administered in overdose may exceed the binding capacity of the plasma protein and lead to an excess of free drug, which can diffuse into various target tissues and produce exaggerated effects. Other reasons the fraction of unbound drug might increase include hypoalbuminemia and competition with other highly protein-bound drugs. The extent of protein binding of a drug cannot be extrapolated between species; in most species, however, NSAIDs, antifungal imidazoles, and doxycycline are examples of highly protein-bound drugs. More rapid clearance of the now unbound drug may mitigate the impact of higher drug concentrations.
Dissociation of a drug from plasma proteins also influences elimination from the body in that drugs more tightly bound tend to have much longer elimination half-lives because they are released gradually from the plasma protein reservoir (eg, cefovecin or long-acting sulfonamides).
Most unbound drugs pass easily from capillaries to extracellular fluid. However, physiological barriers presented by pharmacological sanctuary sites and compartments (eg, blood-brain, placental, and mammary barriers) prevent less-soluble drugs from spreading to all tissues. For the CNS, drugs may gain access through either the capillary circulation (blood-brain barrier) or the CSF (also a blood barrier). Drugs penetrate the cortex more rapidly than the white matter, probably because of the greater delivery rate of drug via the bloodstream to the tissue.
The pharmacological factors and consequences of the diverse rates of entry of different drugs into the CNS are clinically relevant because water-soluble, ionized drugs are less likely to enter the CNS, whereas drugs with low ionization, low plasma-protein binding, and a fairly high lipid-water partition coefficient penetrate more rapidly. Inflammation (eg, meningoencephalitis) can substantially alter the permeability of the blood-brain barrier. In general, direct injection of a drug into the CSF is undesirable because of the risk of unexpected effects.
In pregnant animals, the extent of placentation may determine the extent of a placental barrier. Nutrients such as glucose, amino acids, minerals, and even some vitamins are actively transported across the placenta. Drugs cross the placenta largely by lipid diffusion. The distribution of drugs within the fetus follows essentially the same pattern as in the adult, with some differences regarding the volumes of drug distribution, plasma-protein binding, blood circulation, and greater permeability of interceding membranous barriers. Drugs that are potentially teratogenic should be avoided, particularly in early gestation.
The mammary gland epithelium, like other biological membranes, acts as a lipid barrier, and many drugs readily diffuse from the plasma into milk. The pH of normal milk varies, ranging in goats and cows from 6.5 to 6.8. Therefore, weak bases tend to accumulate in milk. Drugs delivered by intramammary infusion can diffuse into plasma to a greater or lesser extent depending on drug and host factors.
After a drug is distributed into tissues, binding to macromolecules such as protein components of cells or fluids, dissolution in adipose tissue, formation of nondiffusible complexes in tissues such as bone, incorporation into specific storage granules, or binding to selective sites in tissues all impede the movement of drugs back into plasma and account for differences in the cellular and organ distribution of particular drugs.
Drug Metabolism
Metabolism and subsequent excretion of drugs together comprise drug elimination from the body.
Most drug metabolism occurs in the smooth endoplasmic reticulum of the liver. Metabolism generally consists of two phases.
Phase I induces a chemical change (most frequently oxidation, but also reduction) that renders the drug more conducive to phase II. Phase II is a conjugative or synthetic addition of a large, polar molecule that renders the drug water soluble and amenable to renal excretion.
Four possible sequelae follow phase I metabolism: inactivation (eg, most NSAIDs); activation from a pro-drug to the active form of the drug (eg, enalapril to enalaprilat); modification of activity—ie, the formation of active metabolites that may be characterized by activity greater than (eg, tramadol), less than (eg, diazepam), or equal to that of the parent compound; and formation of toxic metabolites, which is generally due to direct cell damage (eg, acetaminophen). In some instances, the toxic metabolite acts as an antigen, causing immune-mediated toxicity (eg, sulfonamides).
Because phase II drug metabolism almost exclusively inactivates drugs (the notable exception being some acetylated and methylated drugs), it often protects the organ of metabolism from drug-induced toxicosis. This is particularly true with the addition of glutathione, which scavenges oxygen radicals; in the face of drug toxicity, N-acetylcysteine will increase the intracellular concentration of glutathione.
Multiple isoforms of phase II drug-metabolizing enzymes exist. Glucuronide (the addition of which is catalyzed by glucuronosyltransferase) is the most common phase II reaction; cats are deficient in some, but not all, glucuronosyltransferases. Other important phase II enzyme-mediated metabolic processes include sulfation, acetylation, and methylation. Species differences in drug metabolism arise from differences in enzyme expression (eg, pigs have a reduced capability for sulfation, and acetylation is deficient in dogs). Amino acid conjugations are particularly important in avian species.
Phase I drug metabolism is largely, but not exclusively, accomplished by heme-containing enzymes referred to as cytochrome P-450 (CYP450) enzymes. Eighteen CYP450 families have been identified in humans, with some being specific for certain drugs or drug classes; others, however, are characterized by broad substrate specificity. The cytochrome family demonstrated to be responsible for most drug metabolism in humans is CYP3A. Others important to drug metabolism are CYP2C and CYP2D. CYP450 enzymes are responsible for synthesis (eg, adrenal steroids, fatty acids) and metabolism of many endogenous compounds.
Drug metabolism is thought to be negligible in early life, and neonates are less able than adults to eliminate lipid-soluble drugs. Therefore, dosing intervals for neonates should be prolonged. Postnatal development in the liver appears to be biphasic, consisting of a rapid and nearly linear increase in activity during the first 3–4 weeks, followed by slower development up to the tenth week after birth. In older animals, decreases in hepatic mass, hepatic blood flow, and drug-metabolizing enzyme activity should lead to longer dosing intervals and, for drugs characterized by first-pass metabolism, to lower oral doses.
Many disease states impair the normal activity of the hepatic drug-metabolizing enzyme systems, in turn decreasing clearance and thus prolonging the half-lives of many drugs. Hepatotoxicity, acute hepatitis, and other extensive liver lesions invariably depress enzyme activity. Evidence of decreased concentrations of plasma albumin as a result of hepatic disease generally indicates that hepatic drug metabolism will likewise be decreased. Drug-metabolizing enzyme activity tends to be decreased by hypothyroidism and increased by hyperthyroidism.
Pharmacogenomics is the study of the genetic basis for differential responses to drugs. Differences in the duration of the action of drugs in various species frequently can be attributed to differences in their rates of biotransformation. Species variations in drug metabolism are common. CYP3A4 is responsible for the broadest substrate activity in humans; however, this may not be true in animals, and it varies among species.
Differences in the content and activity of CYP450 that are determined by age, sex, breed, and species are likely to play a role in differing responses to drugs. Polymorphisms can result in poor metabolizers instead of efficient metabolizers, and decreased metabolism predisposes poor metabolizers to drug toxicosis. Such variants for certain enzymes have been described in humans as being responsible for potentially lethal differences in drug metabolism; they have also been described in dogs (eg, ivermectin toxicity in collies).
Several other factors affect the rate and extent of drug metabolism. Drug interactions may reflect the induction (eg, phenobarbital, rifampin, griseofulvin) or inhibition (eg, chloramphenicol, cimetidine, imidazole antifungals) of CYP450 enzymes, with the impact varying among isoenzymes and the sequelae of drug metabolism. Nutritional state can also affect drug metabolism. The liver is not the only site of CYP450 activity, with sanctuaries and portals of entry being examples of sites where CYP450 activity can alter several aspects of ADME.
In addition to differences in CYP450 enzymes, differential handling of enantiomers is increasingly recognized among species. Enantiomers are mirror images of drug molecules that result when groups of atoms rotate around a central, or chiral, carbon. Generally, such compounds exist as racemic mixtures (50:50 of each isomer); however, the body frequently handles each stereoisomer differentially, with differences also occurring among species. Many cardiac drugs and NSAIDs exist as racemic mixtures of enantiomers, an important consideration when either dosages or withdrawal times are extrapolated from one species to another. Some drugs are enantiopure, containing only as one isomer or the other (eg, levetiracetam, dexmedetomidine, esomeprazole).
Species differences have been better documented for phase II metabolism. Among the more important phase II reactions and the species in which the reactions are deficient are glucuronidation (cats), glutathione transferase (important for scavenging potentially toxic metabolites), sulfation (swine), and acetylation (dogs).
Drug Excretion
Excretion irreversibly removes drugs (or metabolites) from the body.
The kidneys are the principal organ of excretion; however, the liver, GI tract, and lungs also may play important roles. Milk, saliva, and sweat are usually less important; the presence of an active drug in milk, however, may affect nursing young and is a consideration in recommended milk withdrawal times.
Renal excretion of foreign compounds is accomplished either by glomerular filtration, passive diffusion into and out (eg, resorption) of the tubular lumen, and carrier-mediated secretion (eg, active transport or facilitated diffusion). Drugs that are lipid soluble will be passively reabsorbed as urine concentrates in the kidney unless they are converted by enzymatic processes to water-soluble drugs.
Among the more important factors determining renal excretion is renal blood flow, which in turn is influenced by cardiac output. Glomerular filtration in particular, but also carrier-mediated transport, is influenced by changes in cardiac output. Drugs that affect renal blood flow (eg, inhibitors of angiotensin-converting enzyme) can also affect renal clearance.
Only unbound molecules < 66,000 daltons in size are readily filtered through the glomerular membranes into the tubular lumen. Most active tubular secretion of drugs into tubules (renal exertion) occurs in the proximal convoluted tubule. Binding to plasma proteins usually does not hinder the tubular excretion of drugs. Transport proteins exist for weak acids (anions), bases (cations), and organic compounds. Most (passive) reabsorption of nonionized, lipid-soluble drugs occurs as urine is concentrated in the distal and collecting tubules. Acidification or alkalinization of the urine may alter the rate of excretion of some drugs because of ion trapping in the tubular fluid.
Concurrent administration of either acidic or basic drugs that are substrates for carrier-mediated secretion processes prolongs elimination of the drug that has the lesser affinity for the carrier sites, thus increasing its duration of action.
One of the more important factors affecting renal excretion is renal or cardiac disease. Decreased renal blood flow will result in a proportional decrease in renal excretion. However, renal disease may also be accompanied by changes in the metabolic capacity of the kidneys and production of toxins that will compete with the drug for protein-binding sites. Changes in acid-base balance may also influence urine pH.
Drugs and their metabolites may also be excreted either passively or actively by hepatocytes into the bile canaliculi and, ultimately, into the duodenum in the bile (eg, doxycycline). Generally, biliary excretion occurs for drugs with molecular weights > 600 daltons. Biliary excretion is a relatively slow process. Further, many drugs excreted in bile as glucuronide conjugates may become unconjugated by intestinal microflora. Released drug can be reabsorbed into the systemic circulation, resulting in enterohepatic circulation. Such recirculation often accounts for the prolonged half-lives of drugs that are excreted primarily in bile. Impairment of the excretory functions of the hepatocytes or obstruction of bile flow interferes with the biliary excretion of drugs. Dose or interval should then be adjusted accordingly.
Other routes of excretion are less clinically important.
Many drugs are excreted in the feces either because of limited oral absorption or because of diffusion directly into the GI tract.
The ruminoreticulum can act as a drug reservoir because of ion trapping or can remove a drug because of microbial metabolism.
The tracheobronchial tree also may be a potential avenue of excretion, as is the alveolar elimination of inhalation anesthetics. The main factors governing elimination by this route are the same as those determining the uptake of inhalation anesthetics: the concentrations in plasma and alveolar air, and the blood-gas partition coefficient.
The mammary and salivary glands excrete drugs by nonionic passive diffusion. The salivary route of excretion is important in ruminants because they secrete such voluminous amounts of alkaline saliva, although such drugs are likely to enter the GI tract.
For More Information
Mealey KL, Martinez SE, Villarino NF, Court MH. Personalized medicine: going to the dogs? Hum Genet. 2019;138(5):467-481. doi: 10.1007/s00439-019-02020-w. Epub 2019 Apr 28. PMID: 31032534.







