



Pharmacodynamics is the branch of pharmacology concerned with the biochemical and physiological effects of drugs and their mechanisms of action on the body as well as on microorganisms or parasites within or on the body. It considers both drug action, which refers to the initial consequence of a drug-receptor interaction, and drug effect, which refers to the subsequent effects.
By contrast, pharmacokinetics is the branch of pharmacology focusing on the time-course of drug concentrations. However, pharmacokinetics and pharmacodynamics are interrelated in that drug concentrations drive clinical effects.
Drug mechanisms of action can be broadly grouped as protein mediated or non–protein mediated:
Protein-Mediated Mechanisms of Drugs
Drugs or toxins most often target specific proteins including enzymes, ion transporters and channels, receptor proteins, or DNA, or subcellular structures like microtubules. Examples of drug classes (and their protein targets) include the following:
NSAIDs (cyclooxygenase)
Digitalis glycosides (Na+/K+-ATPase)
Local anesthetics (sodium channels)
Proton pump inhibitors (H+/K+-ATPase in gastric parietal cells)
Loop diuretics (Na+/K+/2Cl−-symporter in the thick ascending limb of the renal loop of Henle)
Fluoroquinolones (bacterial DNA gyrase)
Vinca alkaloid drugs like vincristine and vinblastine interact and disrupt microtubules of the mitotic spindle.
Non–Protein-Mediated Mechanisms of Drugs
Although most drugs interact with protein targets (receptors and enzymes), drugs may also exert an effect via physical interactions with body fluids or tissues. Examples of such drugs include the osmotic diuretic mannitol and orally administered antacids, which change the osmolality or pH of bodily fluids and do not directly interact with tissues or organs.
Receptor-Mediated Signal Transduction Mechanisms in Animals
Cellular receptor regulation and signal transduction mechanisms are important to the impact of drugs on patients. Most drugs act via receptors connected to signal transduction mechanisms.
Key characteristics of receptors include the following:
Relative structural specificity for a class of compound
Finite number (ie, receptors are saturable)
Linkage to a distinct biological effect
Receptor specificity is not absolute but rather depends on the binding affinity between a ligand (ie, drug) and its binding site on a receptor. As the dose and concentration of a drug increases, it may increase its interaction with nontarget receptors, leading to adverse clinical effects.
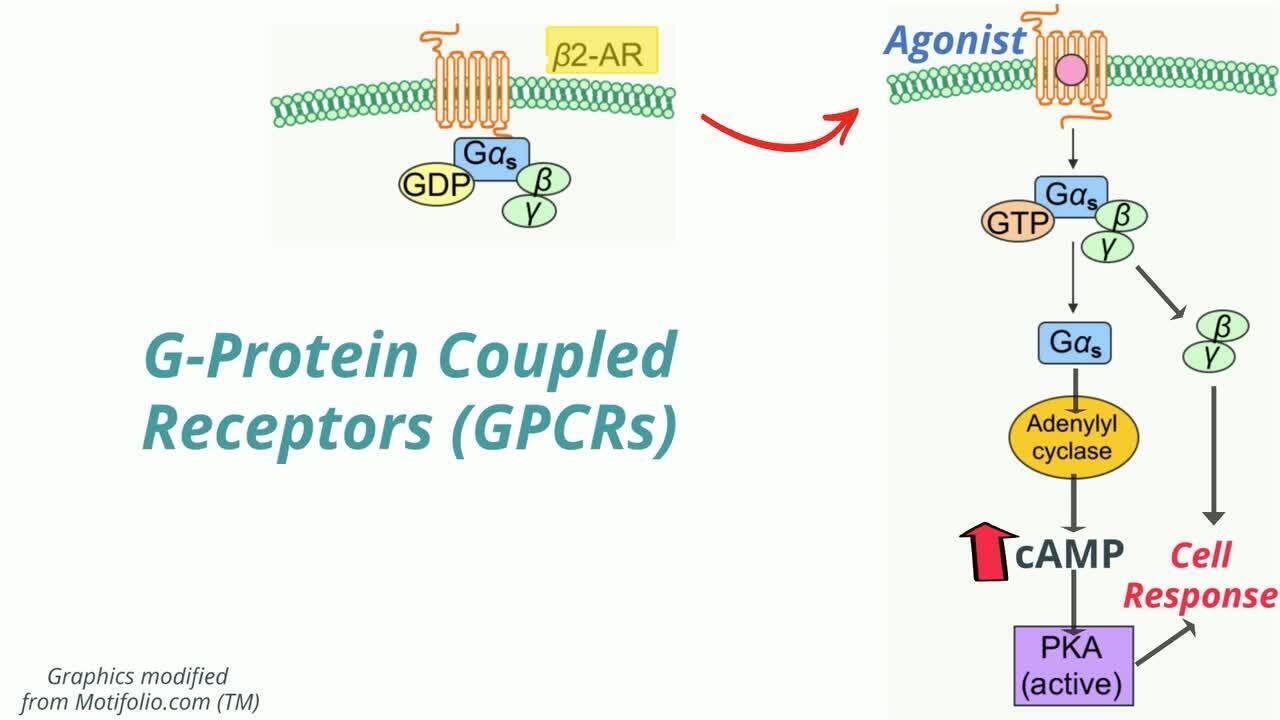
Signal transduction is usually accomplished with one or more key cellular processes, each of which results in allowing a signal (eg, a drug) to convey its effect from outside the cell to the intracellular compartment. There are several categories of signal transduction systems ().


Transmembrane receptors are classified as either ionotropic (linked to ion channels) or metabotropic (linked to biochemical processes). Ionotropic receptors are drug ligand-gated transmembrane ion channels. Examples include gamma-aminobutyric acid (GABA)-linked chloride channels and nicotinic acetylcholine receptors linked to sodium channels.

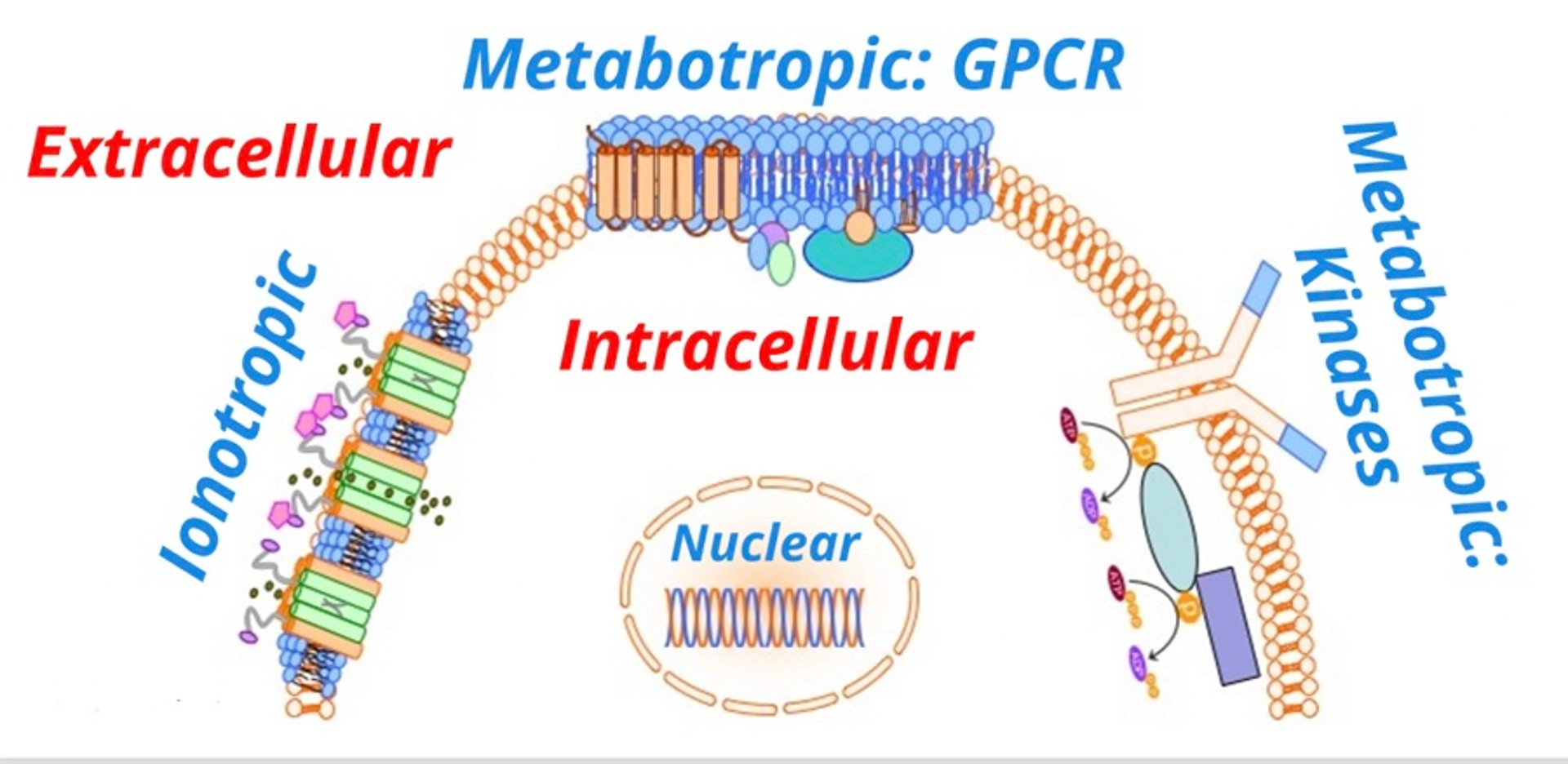
Diagram showing general types of receptor-mediated signal transduction. Transmembrane receptors may be either ionotropic (linked to ion channels) or metabotropic (linked to biochemical processes). Ionotropic receptors are drug ligand-gated transmembrane ion channels. Metabotropic receptors include G-protein-coupled receptors (GPCR) and other protein kinases. Intracellular receptors include cytosolic and nuclear receptors.
Courtesy of Dr. Duncan C. Ferguson.
Metabotropic receptors act directly or indirectly on signal transduction enzymes or are linked to enzymes that have an extracellular domain recognizing a drug and an intracellular domain that catalyzes a biochemical response. This receptor subtype includes G-protein-coupled receptors (GPCRs).
Other receptors are guanylyl cyclase (GC)-linked. In this case, the plasma membrane receptors have guanylate cyclase activity and the cyclic GMP formed activates protein kinase G (PKG), which phosphorylates proteins associated with cellular action. Examples of this type of receptor include receptors for atrial natriuretic peptide (ANP) and nitric oxide (directly or via muscarinic receptors).
Other receptors are those linked to kinase enzymes, such as the insulin receptor, which has intrinsic tyrosine kinase enzyme activity and can transfer a phosphate group from ATP to a protein (in this case, insulin receptor substrate 1 [IRS-1]) in a cell. This activates additional proteins via phosphorylation, leading to the translocation of glucose by glucose transporter type 4 (GLUT4). Receptors of this type can be regulated in a variety of ways via phosphorylation and dephosphorylation.
Finally, there are intracellular receptors, including cytosolic receptors and nuclear receptors. Examples of nuclear receptors include those for lipophilic hormones, such as thyroid and steroid hormones, which bind to a DNA response element, resulting in inhibition or activation of mRNA and, finally, protein transcription.
Drug-Receptor Binding in Animals
Pharmacokinetics predicts that plasma (and tissue) concentrations are proportional to the administered dose. Likewise, the amount of drug bound to receptor is also usually proportional to the rate of signal transduction (ie, effect). However, the intermediate step of receptor binding adds a nonlinear component so that, overall, the drug response or effect is nonlinearly related to drug concentration and dose.
Most drugs interact with receptors in a dynamic equilibrium, meaning that there is reversible binding to the receptor. Due to this reversibility, the plasma concentration drives the drug response in a proportional manner until the maximal effect is achieved, often, but not always, when all receptors are occupied. ().

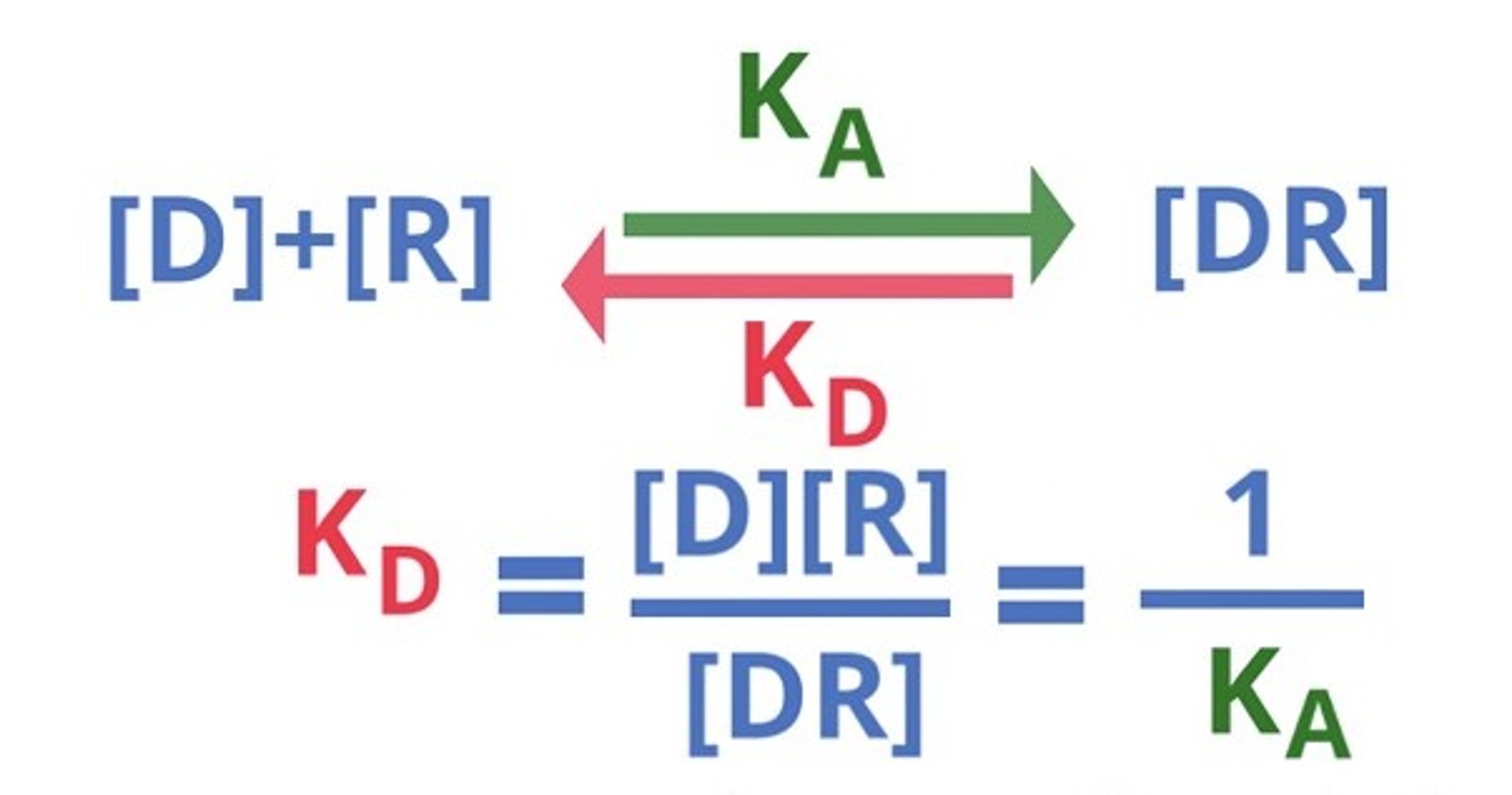
Diagram showing the dynamic equilibrium of drug-receptor interaction. [D] = Drug concentration. KD = Dissociation constant. [R] = Receptor concentration. KA = Association constant. [DR] = Drug-receptor complex concentration.
Courtesy of Dr. Duncan C. Ferguson.

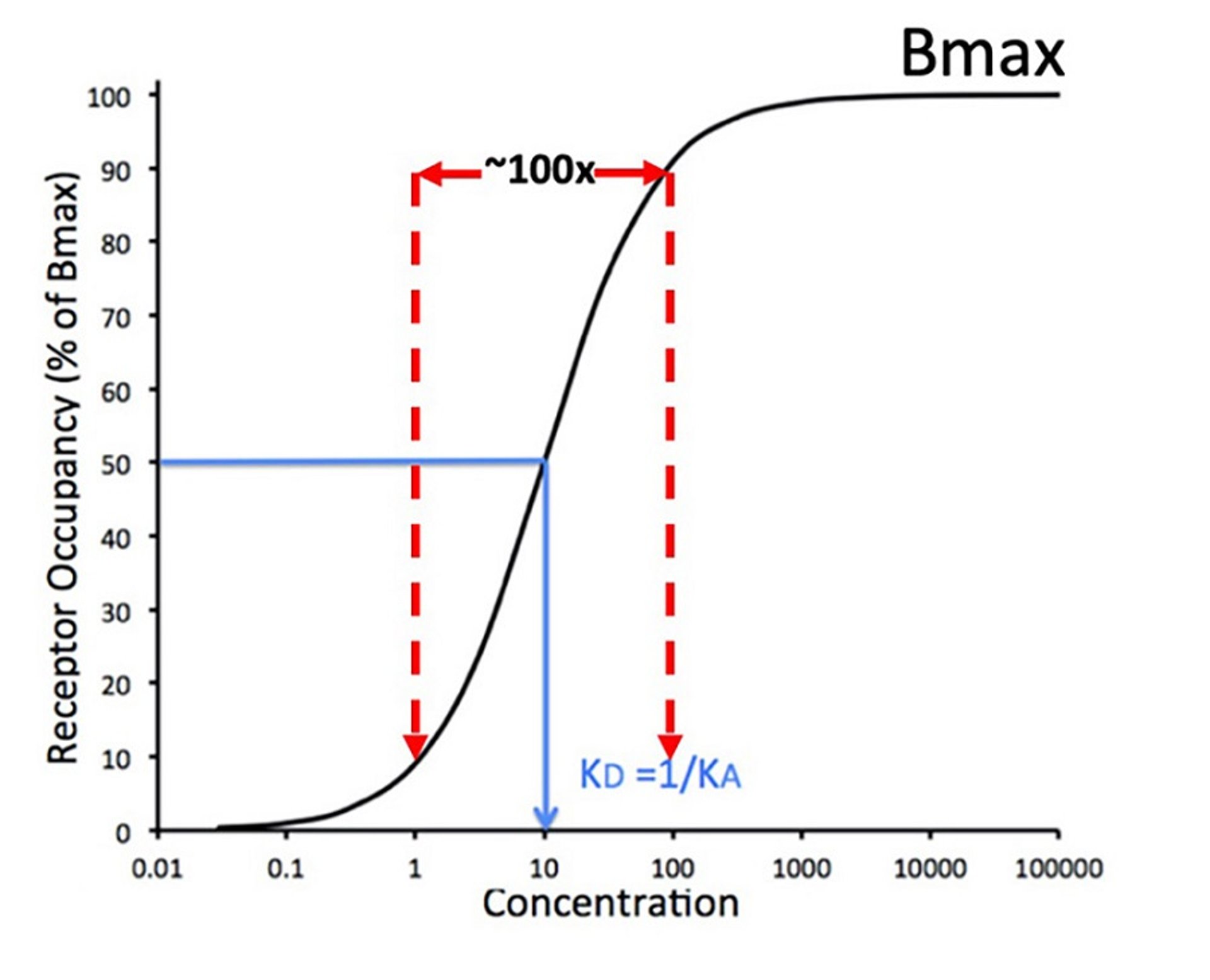
Dose-response curve for drug-receptor binding showing receptor occupancy vs drug concentration (on a logarithmic scale). Mass action of binding predicts a nonlinear dose-response curve, which is hyperbolic but often shown as a sigmoidal plot. Bmax = Maximal binding capacity.
Courtesy of Dr. Duncan C. Ferguson.

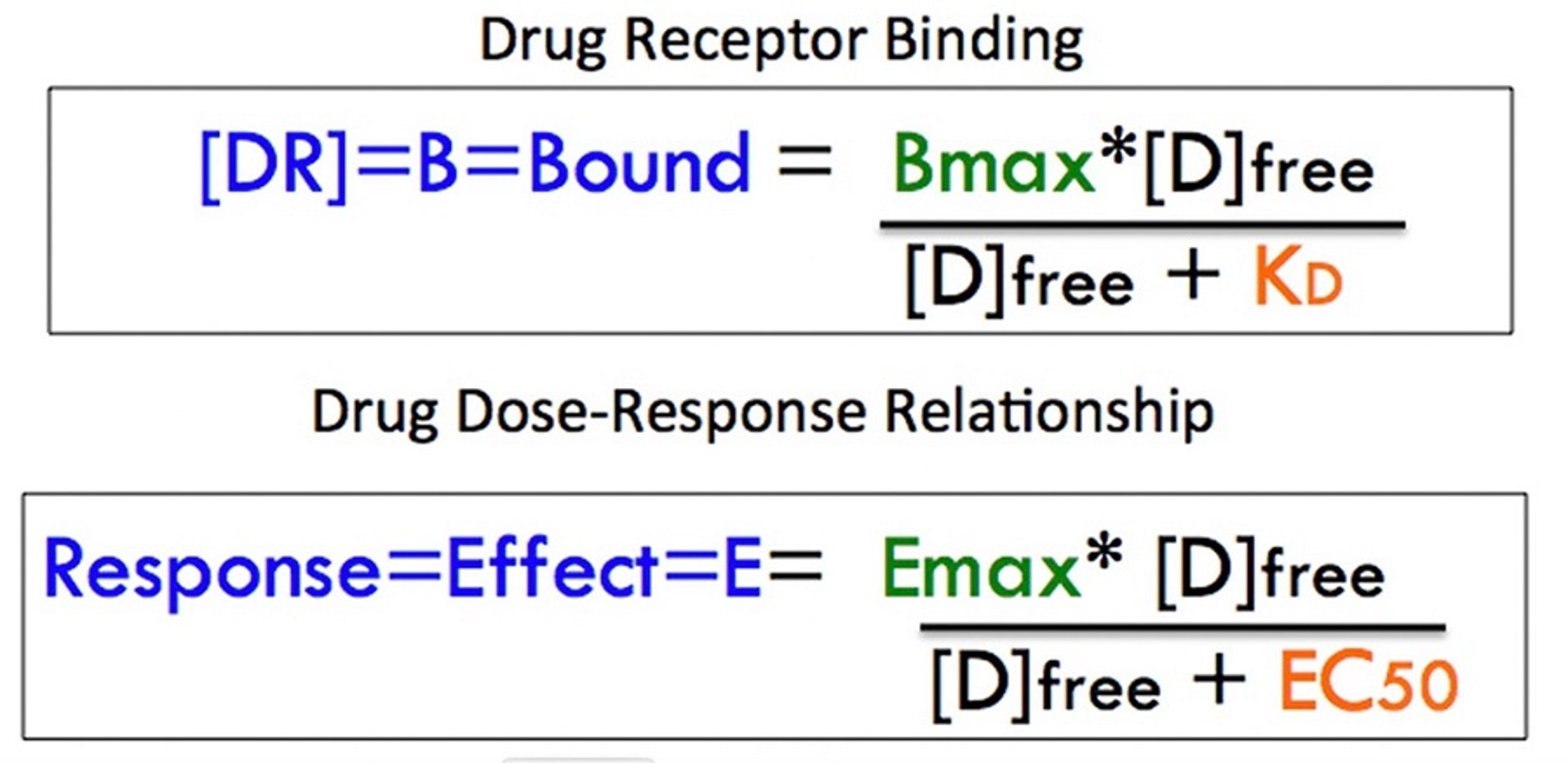
Diagram comparing drug-receptor binding to drug dose-response relationship. A comparison with the curve defined by drug binding shows that the drug effect is described by a similar equation in which maximal binding (Bmax) is an analogous term to maximal effect (Emax) and the dissociation equilibrium constant KD is replaced by the median effective concentration (EC50). [D]free = Concentration of drug not bound to receptor.
Courtesy of Dr. Duncan C. Ferguson.
However, mass action of binding predicts a nonlinear dose response curve, which is hyperbolic but often shown as a sigmoidal plot of the log-dose (concentration) vs response curve (). The dissociation equilibrium constant (KD) defines the middle of the operating range of concentration for the drug-receptor interaction, meaning that it is the concentration at which 50% of the receptors are occupied. In general, a drug will show its full range of effect within ~ a 100-fold concentration range. This is exemplified in : the drug's threshold activity occurs at about a concentration of 1 and approaches maximal binding at a concentration of 100, with 50% of maximum achieved at a concentration of 10. The maximum binding capacity (Bmax) is representative of the finite amount of receptor protein present in a cell or subcellular fraction.
Log-Dose Effect Relationship in Animals
Because the drug concentration is linearly related to the dosage, the relationship between drug concentration and effect can be described with a similar equation. ().
A comparison with the curve defined by drug binding shows that the drug effect is described by a similar equation in which maximal binding (Bmax) is an analogous term to maximal effect (Emax) and the dissociation equilibrium constant KD is replaced by the median effective concentration (EC50).
Receptor Agonists in Animals
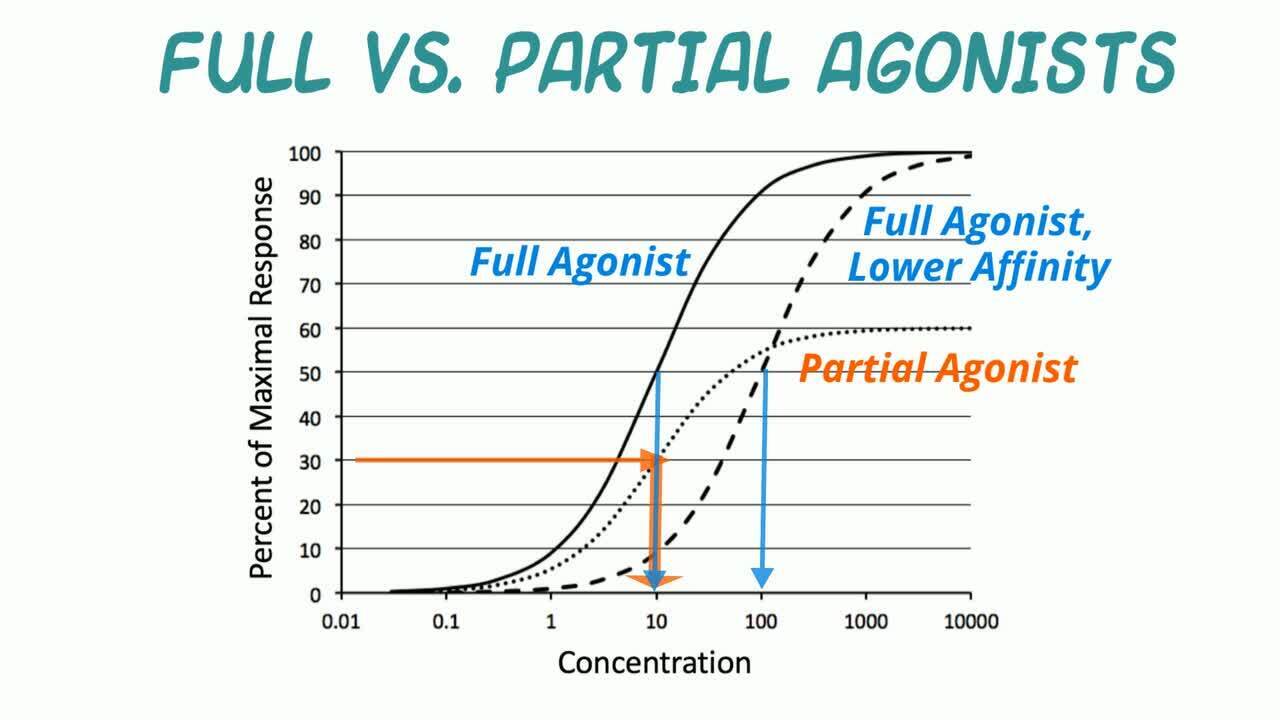
Drugs that drive a signaling pathway forward, leading to a biological effect often associated with a therapeutic effect, are called agonists.
Compounds interacting with receptors are divided into distinct pharmacological types based on their intrinsic efficacy on a receptor-signal transduction system. A full agonist achieves the largest biological effect at its maximally effective concentration. A partial agonist is an agonist for which the Emax, and therefore the intrinsic efficacy, is less than that of a full agonist. ().

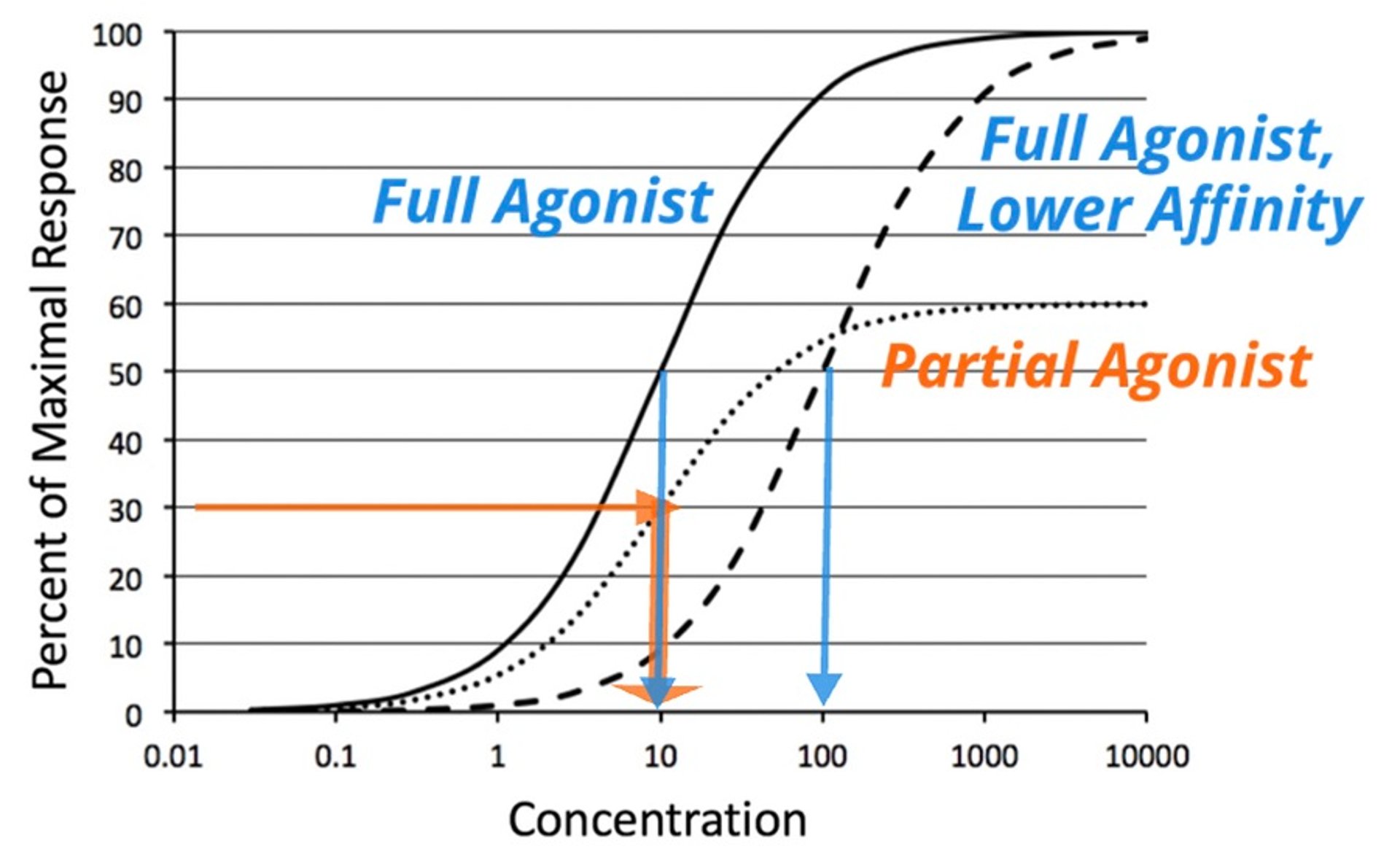
Log-concentration vs response curves for full agonists with 2 affinities and a partial agonist. The solid line traces the effect of a full agonist. Because the same maximal response is achieved, the drug represented by the dashed line is also a full agonist; however, it has a lower potency (presumably due to decreased receptor binding affinity). The dotted line shows the curve for a partial agonist. Notice that it has a lower Emax than the other 2 drugs. Also, the EC50 of this curve shows that this partial agonist has the same potency (EC50 = 10) as the full agonist shown in the solid line but a higher potency than the full agonist shown with the dashed line (EC50 = 100).
Courtesy of Dr. Duncan C. Ferguson.
A classic example of a partial agonist is buprenorphine, a mu-opioid partial agonist. Its activity is compared with morphine, a full mu agonist. However, buprenorphine is about 30-fold more potent than morphine because it binds to the receptor with higher affinity. The clinical benefit of a partial agonist would be associated with the safety of the ceiling for its effect (ie, its Emax). A lower Emax would be associated with a lower tendency for the adverse effect of respiratory depression with opioid drugs.
Understanding Drug-Receptor Transduction in Animals
When two drugs bind to the same receptor connected to the same signal transduction system but lead to different maximal effects, they have different intrinsic efficacy. A receptor can be in two states: the resting state (R) and the activated state (R*). These states are in equilibrium with each other but can be impacted by the type of drug that binds to them.
When a full agonist interacts with the receptor, it shifts the equilibrium toward the activated receptor form, and it has a preferential affinity for the activated form relative to the resting form. The greater the conformational selectivity for R*, the greater the efficacy. With a partial agonist, the maximal effect is less (ie, partial) than that of a pure agonist. Considering receptor theory, the partial agonist, although favoring R*, does not shift the conformational equilibrium toward R* as effectively as a pure agonist. So even at maximal effect, the effect is less than the maximal effect for a full agonist.
A useful analogy is to compare a drug's effect to that of a bicycle driven by a pedal and chain with multiple gears. In this analogy, the same number of rotations of the pedals (ie, concentration and receptor affinity) of a full agonist would move the bicycle farther down the road than a partial agonist (ie, the Emax for a full agonist is greater than the Emax for a partial agonist). To complete the analogy, a low gear would relate to a low affinity agonist and a higher gear would relate to a high affinity agonist, affecting mainly the speed achieved by the bicycle (effect) at a given rate of pedaling (concentration), and not the limit of the distance traveled (Emax).
Although it might seem counterintuitive to use a drug that can have only part of the maximal effect, this may be desirable in the case of some drugs, such as opioid agonists. Morphine and oxymorphone, for example, are considered pure or full agonists. However, their dose-dependent beneficial effects and adverse effects (eg, suppression of the respiratory center, which can lead to death) overlap considerably, making their administration quite dangerous. However, the partial opioid agonists like butorphanol or buprenorphine can be administered more safely because their beneficial effects level out before reaching the point where the adverse effect of respiratory depression is typically observed.
Antagonists in Animals
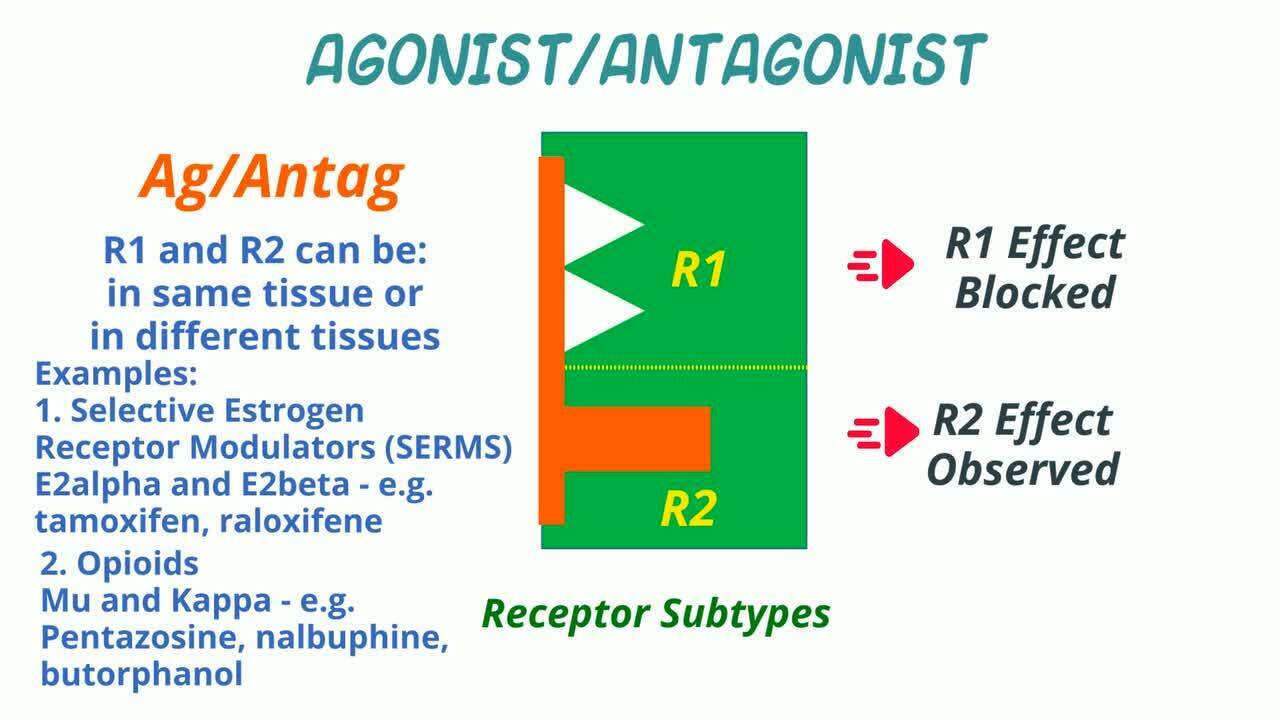
Drugs that tend to act on receptors to diminish certain signaling pathways are known as antagonists, agonist/antagonists, and inverse agonists. Receptor antagonist drugs block the action of an endogenous or exogenous compound, either via direct competition at the receptor or via alteration of the receptor function.
Most receptor antagonist drugs are considered competitive antagonists, which means that they compete with an agonist for binding to a receptor by mass action but do not alter the receptor. From a clinical perspective, pure antagonists have no intrinsic efficacy of their own, but compete with the agonist based on their receptor affinity (usually designed to be greater). However, their effect only occurs by blocking the effect of an endogenous or exogenous agonist (pure or partial).
Other terms used for antagonists are "blockers" and "-lytic" drugs. For example, the beta1-receptor antagonist atenolol is often referred to as a beta blocker and is considered a sympatholytic drug.

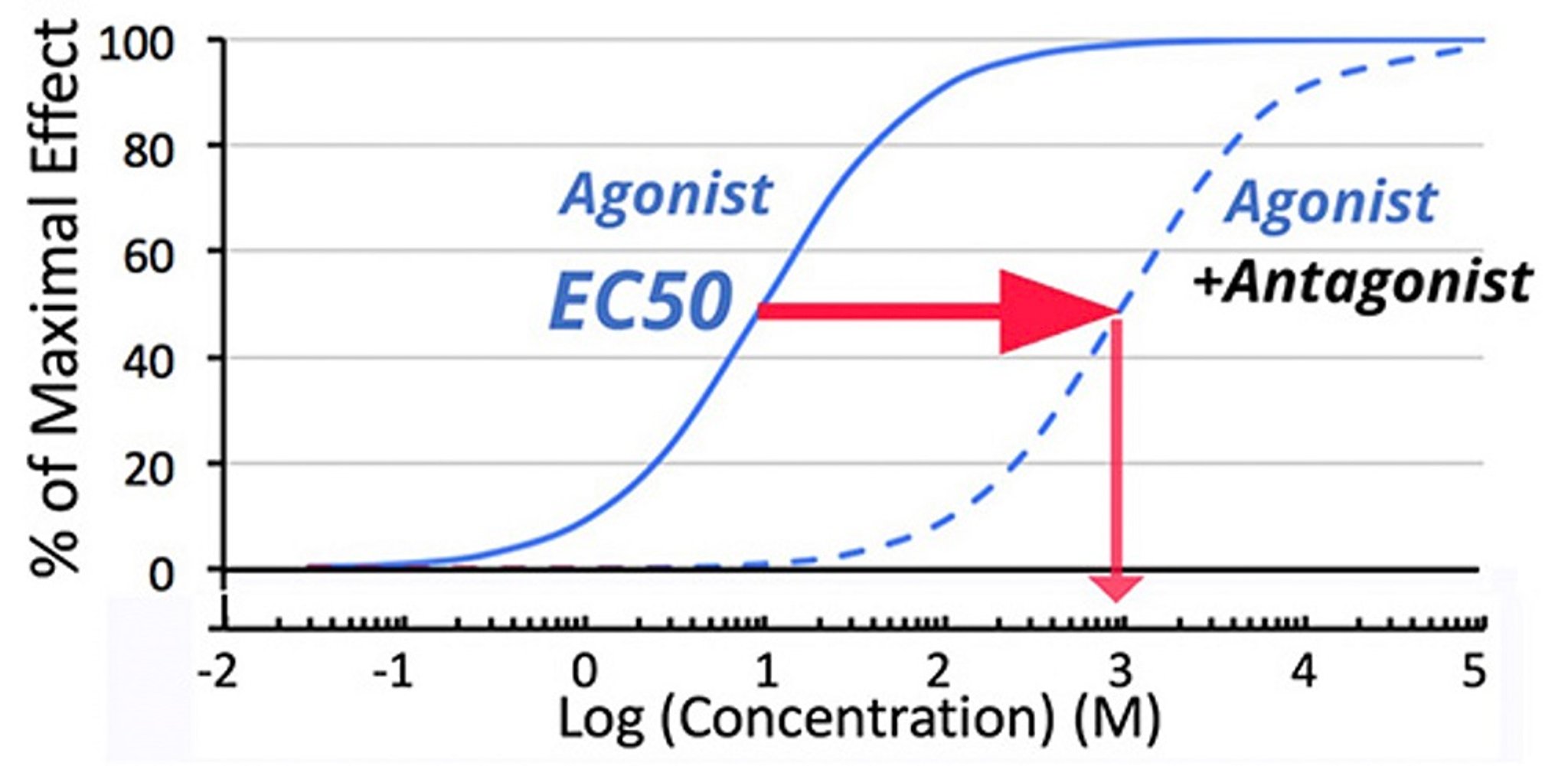
Dose-response curve for an agonist drug with and without the presence of a competitive antagonist. In the presence of a competitive antagonist, achievement of the same effect as originally achieved by the EC50 of agonist by itself requires an agonist concentration that is 100-fold higher (indicated by arrows).
Courtesy of Dr. Duncan C. Ferguson.
A log concentration vs effect plot ( See figure Dosages of Antifungals for Respiratory Therapy) shows that the addition of a competitive antagonist shifts the dose-response curve for an agonist to the right, meaning the EC50 will be an apparently higher in the presence of a competitive antagonist. Although not all competitive antagonists have this high affinity, when they do and concentrations of antagonist are maintained, they almost appear irreversible because the available agonists cannot be administered in adequate amounts to compete with the antagonist. More importantly for the veterinarian, the effect that occurs at the original EC50 is miniscule, leading to the clinical effect of blockade.
Conceptualizing this with receptor theory, a competitive antagonist is a compound that binds with equal affinity to receptors in their resting state (R) and activated state (R*). It will compete with a full or partial agonist for receptor binding, but it does not shift the conformational equilibrium and so will lead to no effect on its own.
In the bicycle gear analogy, blockade by a competitive antagonist would be similar to a situation where the chain has come off of the sprocket. Attempts to pedal the bicycle produce no forward movement (ie, due to presence of the competitive antagonist, a normally effective concentration of an agonist no longer leads to a biological effect).
Noncompetitive Antagonists in Animals
A small number of drugs are considered noncompetitive antagonists. These drugs may result in covalent modification of the receptor, eliminating its ability to bind the agonist, or may interact with an allosteric site near the receptor, changing the configuration of the receptor. Either way, noncompetitive antagonists decrease Emax, and no concentration of agonist is capable of overcoming this effect. ().

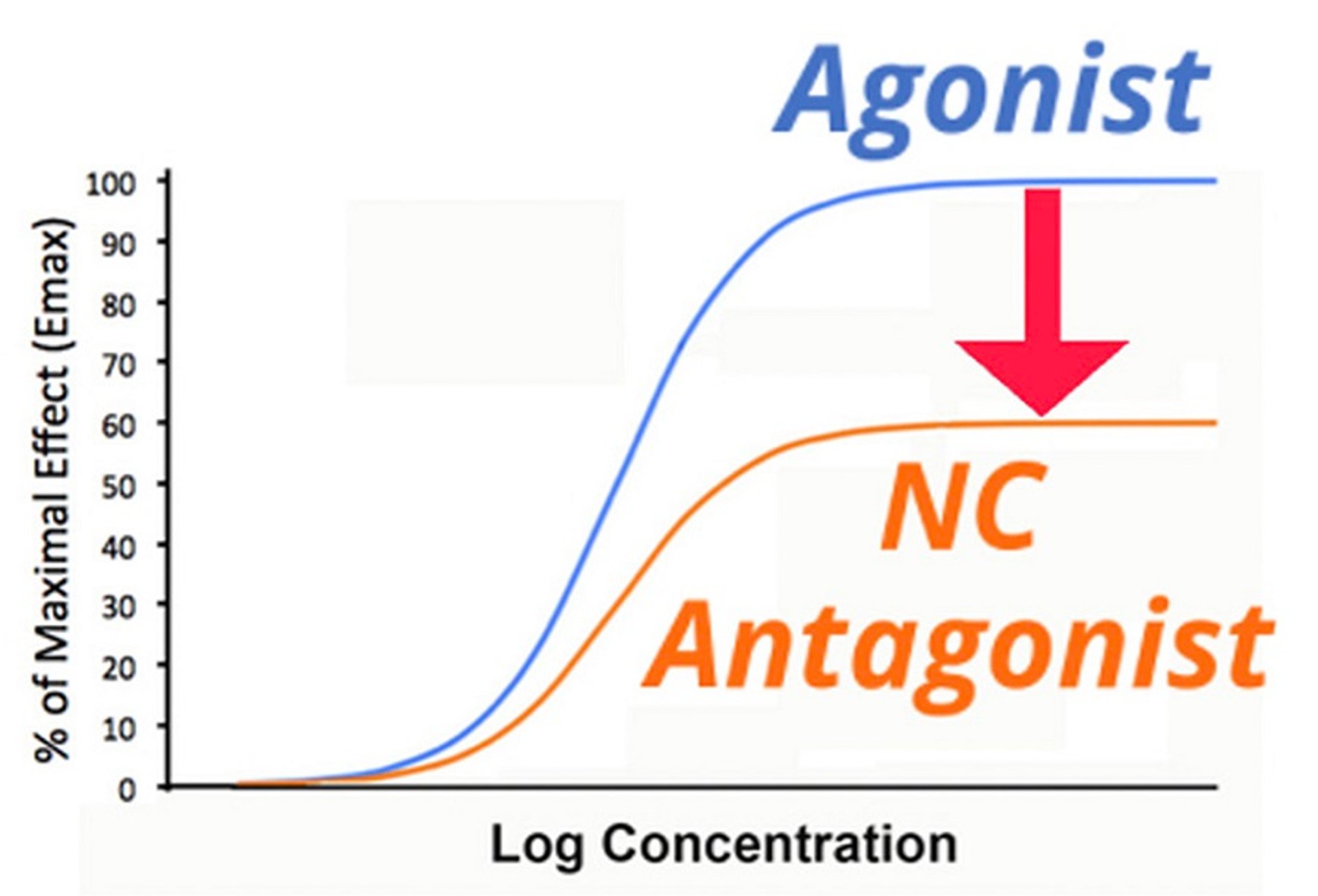
Dose-response curve for an agonist drug with and without the presence of a noncompetitive (NC) antagonist. In the presence of a noncompetitive antagonist, the apparent maximal effect (Emax) of an agonist is decreased (indicated by arrows).
Courtesy of Dr. Duncan C. Ferguson.
An example of this type of drug used in veterinary medicine is the alpha-adrenergic receptor antagonist phenoxybenzamine, which is used most commonly for postcatheterization urinary spasms. This drug interacts with the receptor and after a period of time covalently binds to it. The clinical relevance is that this drug takes some time to have its maximal action. However, the maximal effect that can be achieved by an agonist is decreased, just like with a lowering of receptor numbers (lowered Bmax). Only synthesis of new receptors can return the maximal effect to pre-antagonist levels.
Agonist/Antagonists in Animals
An agonist/antagonist is a drug which is an agonist under some conditions and an antagonist under others (other receptor or receptor subtype or other tissue). In its role as an antagonist, it can block the activity of other agonists. When acting differently depending on the tissue, it might be called a selective receptor modulator. Selective estrogen receptor modulators (SERMs) like tamoxifen or raloxifene are examples.
In veterinary medicine, a prominent example of agonist-antagonists are opioids. Drugs that act as agonists at the kappa-opioid receptor and antagonists at the mu-opioid receptor include pentazocine and nalbuphine. Butorphanol is also a pure kappa-opioid receptor agonist, a partial mu-opioid receptor agonist, and an antagonist at the delta-opioid receptor.
Inverse Agonists in Animals
Drugs can also counter a signal transduction pathway by interacting directly with the transduction system. There are receptors that are associated with intrinsic transduction even when there is no receptor occupancy. Inverse agonists interact with the signal transduction pathway but decrease the pathway's intrinsic activity. ().
Conceptualizing this with receptor theory, an inverse agonist binds with greater affinity to receptors in their resting state (R) than to receptors in their activated state (R*), shifting the conformational equilibrium toward R, the form of receptor which is less effective at driving the signal transduction pathways forward. When interacting with a system where the unliganded receptor has constitutive activity, an inverse agonist serves to decrease the effect below constitutive levels (ie, the dose-response curve is the inverse of that of a full or partial agonist).

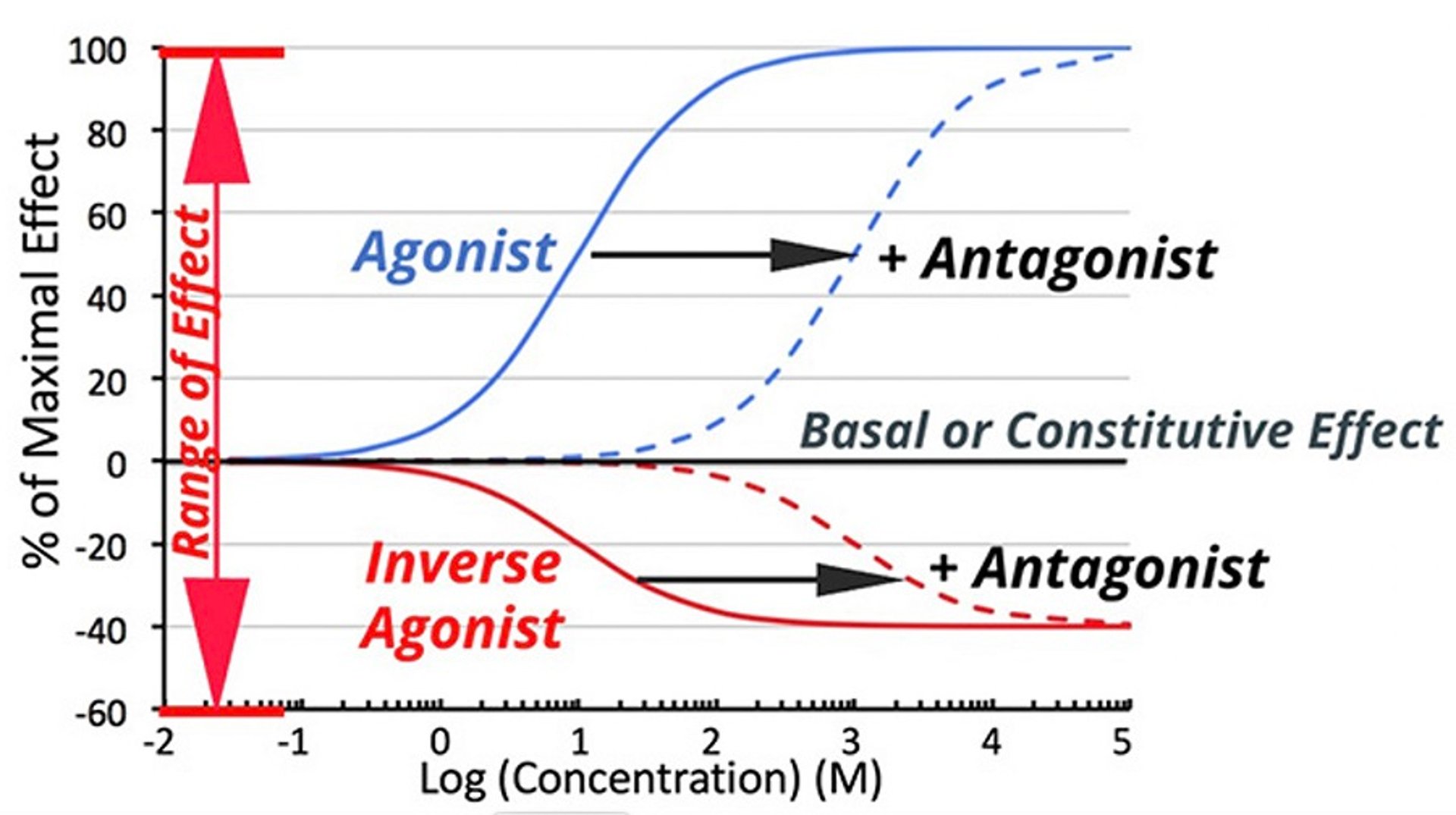
Dose-response curve for inverse agonists compared to full agonists, with and without the presence of competitive antagonists. Note that inverse agonists may also be antagonized.
Courtesy of Dr. Duncan C. Ferguson.
In the bicycle and gear analogy, the inverse agonist would cause the bicycle to go in the direction opposite of a pure agonist. It might also be imagined as if the bicycle was going downhill (intrinsic agonist-free activity) which can be slowed by braking (activity reduced in the presence of an inverse agonist).
Receptor Phenomena of Clinical Relevance in Animals
Response to a given drug can vary between patients. Some patients consistently respond more to a drug and some respond less. Changes can also occur to drug response over time in the same patient because drug receptor amount, affinity, and signal transduction activity vary with time.
Hyporeactivity or Tolerance
The body has multiple mechanisms by which it can adapt to the administration of multiple doses of a drug or toxin, and both receptor and signal transduction mechanisms can be involved with this adaptation. The general term for developed hyporeactivity is tolerance.
The mechanisms leading to tolerance after chronic dosing may be difficult to distinguish in the individual patient unless a complete dose-response study is performed.
Rapid Loss of Drug Effect: Tachyphylaxis
Tachyphylaxis refers to the relatively rapid loss of effect over a short timeframe (minutes to days). For example, decongestant nasal sprays and eye drops often include alpha adrenergic receptor agonists (eg, phenylephrine) for topical decongestion by nasal mucosal blood vessel vasoconstriction. Prolonged (greater than several days) use of a nasal decongestant can lead to a diminution of effect over time. The mechanism of this tachyphylaxis is alpha-adrenoceptor mediated downregulation (reduction) and drug-induced desensitization of response (see below).
Loss of Drug Effect: Receptor Desensitization and Downregulation, and Exhaustion of Mediators
The response of a drug stimulating G-protein coupled receptors (GPCRs) may fall rapidly or more slowly. Exemplified by the beta-adrenergic receptor, rapid desensitization is usually associated with receptor phosphorylation. In a desensitized state, the dose-effect curve would be shifted in parallel to higher concentrations (as seen with competitive antagonists in and ), and the apparent EC50 would be higher, meaning that the same drug dosage (concentration) would lead to a lower effect than initially. When the agonist is removed, cellular phosphatases reverse the effect.
The second mechanism by which a cell or tissue can modulate a drug's action is downregulation, the reduction of receptor numbers. On a log concentration vs effect plot, loss of receptors appears as shown in . With some G-protein coupled receptors, including beta adrenergic receptors, chronic stimulation leads to increasing phosphorylation of the receptor, which then traffics it to lysosomes that destroy the phosphorylated receptor, effectively reducing the number of receptors available on the plasma membrane. Such downregulation is characterized by a lower Emax, a secondary effect of decreased receptor numbers (Bmax).
A third mechanism for reduction of a drug's effects is the exhaustion of signal transduction mediators. For example, the adrenergic agent amphetamine leads to the release of synaptic catecholamines. Chronic administration may lead to the eventual depletion of catecholamine stores.
Hyperreactivity
Increased reaction to a drug may occur when there is a reduction of endogenous physiological receptor ligands, which can lead to supersensitization (associated with a decreased EC50 or increased Emax). For example, beta-adrenergic antagonists such as propranolol or atenolol, used to manage heart rate and blood pressure, decrease signaling via the linked adenylate cyclase. The reduction in cAMP and phosphorylation eventually results in recruitment and upregulation of beta-receptors on the cell membrane. Withdrawal of these drugs would initially be associated with hyperreactivity to endogenous mediators (eg, epinephrine), leading potentially to rebound hypertension. The clinical effect is known as overshoot and is the reason that gradual withdrawal of autonomic drugs is recommended whenever possible to allow cell and tissue responsiveness to return to normal.
Spare Receptors
Spare receptor capacity provides a mechanism for obtaining maximal response at a very low concentration of agonist despite a relatively low affinity (ie, high KD) for the receptor.

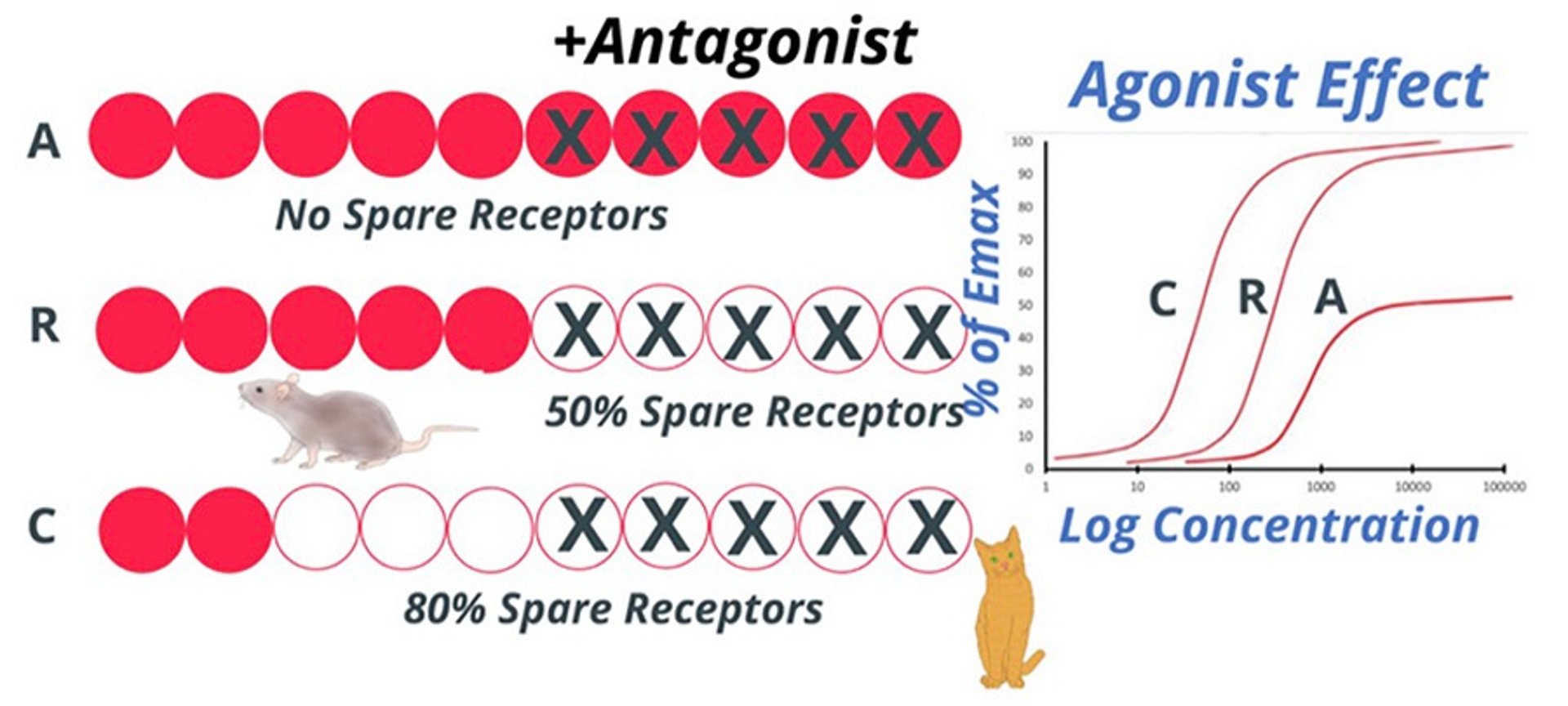
Diagram comparing dose-response curves for an agonist drug in different species with varying levels of spare receptors. A represents a receptor system requiring full receptor occupancy for full effect (the 10 circles represent all of the receptors [red = bound by agonist; white = unbound by agonist; X = bound by antagonist). R (rodent) and C (cat) represent receptor systems in which 50% and 20% occupancy, respectively, can achieve the maximal effect (ie, there are spare receptors). Conversely, in the presence of a competitive antagonist, similar receptor occupancy can lead to reduction of the effect in A but not R or C.
Courtesy of Dr. Duncan C. Ferguson.
With some drugs, the maximal biological effect can be achieved at much lower doses or concentration than expected depending on the receptor's binding affinity. Instead of a change in a patient, this phenomenon is a built-in feature of the receptor system; however, it can vary considerably between species (see figure "Example of spare receptors").
A classic example of spare receptors is found in the nicotinic acetylcholine receptor system. These low-affinity receptors are linked to sodium ion channels. The low affinity of these receptors allows synaptic acetylcholine (ACh), normally at a high concentration of 1 mmol/L, to rapidly dissociate from its receptor to end the initiating neurotransmitter stimulus. The ACh is then destroyed by synaptic acetylcholinesterase. Once depolarized, these receptors undergo temporary desensitization. As a result, to maintain the potential for repetitive stimulation at normal physiological rates, additional receptors must be recruited to maintain contraction.
A clinical scenario illustrating the importance of spare receptors is the administration of d-tubocurarine, which competitively blocks nicotinic receptors in the postsynaptic plasma membrane of skeletal muscle and is used during anesthetic protocols to optimize muscle relaxation. As animals are normally endotracheally intubated during neuromuscular blockade, a predictable and reasonable duration of action is important. For the skeletal muscle relaxation effect, the competitive antagonist d-tubocurarine must not only block the active receptors, but also the spare receptors.
Key Points
Most drugs act via interaction with cellular proteins: enzymes, transporters, or receptors.
The pharmacodynamic effects of drug-receptor interactions are most often reversible in nature and defined by the drug concentration and the equilibrium binding constant.
Drug-receptor binding relationship can be represented as a sigmoidal log dose-response curve.
Drugs that act on receptors to drive a signaling pathway forward are full or partial agonists and those that act on receptors to diminish signaling pathways are antagonists, agonist/antagonists, or inverse agonists.
Response to a given drug can vary between patients depending on drug receptor amount, affinity, and signal transduction activity.
For More Information
Ferguson, DC. Principles of Pharmacodynamics and Toxicodynamics. In Haschek and Rousseaux's Handbook of Toxicologic Pathology. Elsevier Inc.; 2013: 61–76. https://doi.org/10.1016/B978-0-12-415759-0.00003-0
Also see pet health content regarding drugs and vaccines.





